

1 Introduction
Les encéphalopathies spongiformes transmissibles (EST) se manifestent par une neurodégénérescence spongiforme du systèmes nerveux central et affectent aussi bien les animaux que les hommes 〚1〛. Lˈévènement pathologique central de ces maladies semble être la conversion dˈune protéine appelée PrPC, pour forme cellulaire de la protéine prion, vers une isoforme pathologique PrPSc, pour forme ‘scrapie’ de la PrP 〚2〛. Il existe encore de nombreuses incertitudes concernant la fonction normale de la PrPC et les mécanismes impliqués dans la neurodégénérescence et la transmission des EST. D’une façon générale, les cultures cellulaires facilitent grandement l’étude de protéines d’intérêt en permettant l’utilisation de techniques microscopiques et biochimiques très variées. Dans le cadre des EST, ces modèles ont permis des avancés significatives de nos connaissances que nous rapportons dans cette revue.
2 Principaux modèles en culture cellulaire utilisés dans le cadre de l’étude des EST
2.1 Cultures cellulaires classiques et transfectées avec de la PrP sauvage
L’identification d’une protéine, appelée PrPSc, dans les fractions infectieuses responsables des EST 〚2〛 a été une étape déterminante dans l’étude et la compréhension de ces maladies. Il a été rapidement établi que la PrPSc était une forme anormale d’une protéine cellulaire physiologique, la PrPC. Cette dernière est exprimée de façon quasi ubiquitaire dans l’organisme 〚3〛 et par un grand nombre de cultures cellulaires 〚4–6〛. C’est dans les cellules neuronales que la PrP est la plus fortement exprimée et que les études les plus détaillées de la biologie cellulaire de la PrPC et de la PrPSc ont été réalisées. Afin d’augmenter artificiellement la quantité de PrP exprimée en culture et de faciliter la détection et la purification de la protéine, plusieurs groupes ont développé des lignées qui sur-expriment la PrPC après transfection 〚7–10〛.
2.2 Cultures cellulaires transfectées avec des PrP modifiées
La description de mutations de la PrP associées aux formes génétique d’EST 〚11〛 a conduit logiquement à s’intéresser à l’expression de ces PrP mutées dans des systèmes en cultures cellulaires 〚6〛. Ce n’est en fait qu’en 1995 qu’un modèle dans lequel les PrP mutées présentaient des propriétés biochimiques anormales a été établi 〚7〛 (voir figure 1). Ces cultures ont permis d’étudier en détail les mécanismes pathologiques liés aux mutations 〚8, 12–15〛. Parallèlement à ces modèles des formes génétiques des EST, des PrP chimériques et tronquées ont été exprimées en culture cellulaire dans des buts variés d’étude de la fonction ou de la conversion de la protéine 〚16, 17〛.
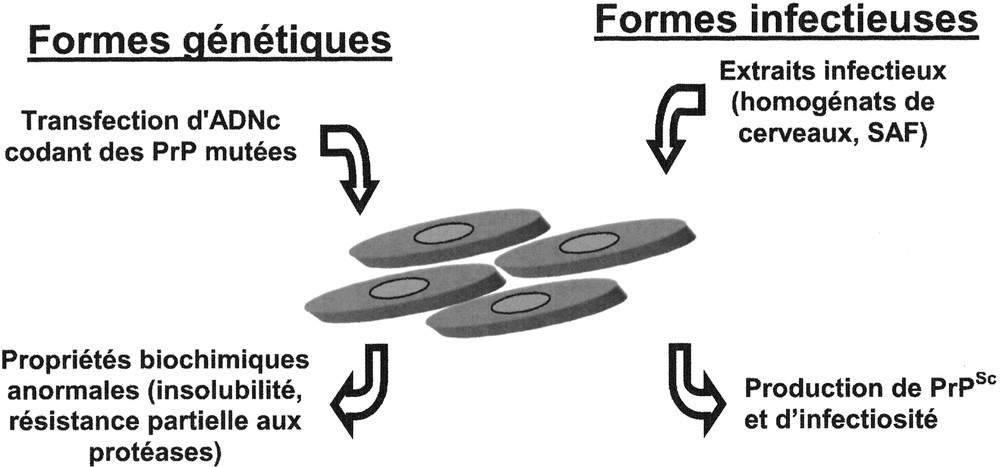
Modèles cellulaires des formes génétiques et infectieuses des EST. Les PrP portant des mutations homologues à celles présentes chez les patients atteints des formes génétiques d’EST acquièrent après transfection en culture cellulaire (partie droite de la figure), des propriétés biochimiques anormales. Afin de générer des lignées cellulaires chroniquement infectées par les prions (partie gauche de la figure), les cultures sont mises en contact avec des extraits infectieux et la présence de PrPSc et d’infectiosité est testée après repiquage successif.
2.3 Cultures cellulaires issues de souris transgéniques
L’obtention de souris transgéniques dénuées du gène codant la PrP (PrP –/– ) 〚18〛 a fait date en montrant que cette protéine était nécessaire au développement et à la transmission des EST. Par ailleurs, la création de souris sur-exprimant la PrP sauvage et ayant une durée d’incubation réduite 〚19〛, de même que celle de souris sur-exprimant une PrP mutée 〚20〛 et tombant spontanément malades ont également été des découvertes essentielles. De nombreuses autres lignées de souris transgéniques produisant des PrP modifiées ont été établies depuis 〚19〛. Ces modèles transgéniques ont permis de dériver des cultures primaires exprimant différents niveaux de PrP et/ou des PrP mutées. Nous citerons en particulier des lignées neuronales issues de souris PrP –/– qui ont été ensuite immortalisées 〚21〛.
2.4 Cultures cellulaires infectées
Historiquement, la propagation de l’agent infectieux des EST en culture a été testée dès 1970 〚22〛. Ces travaux ont été poursuivi par Rubenstein en 1984 〚23〛, puis surtout par le groupe de Chesebro 〚24〛 et de Prusiner 〚25〛 notamment. Le principe de la génération de ces lignées repose sur la mise en contact de cellules avec des extraits infectieux bruts (homogénats 5 de cerveaux) ou purifiés (SAF, ‘scrapie associated fibrills’) (voir figure 1). La présence d’infectiosité testée par inoculation à l’animal permet de confirmer la réussite de l’infection, de même que la détection biochimique de la PrPSc qui est plus rapide et plus simple à mettre en oeuvre 〚26〛. En 1997, Schatzl décrivait une nouvelle lignée cellulaire sensible aux prions 〚27〛 mais le faible nombre de lignées cellulaires infectées, ainsi que la restriction des résultats positifs à certaines souches adaptées à la souris 〚28〛, montrent que de nombreux paramètres limitent la multiplication de l’agent en culture. La quantité de PrP générée de même que la similitude des PrP produites par les cultures et dans l’inoculum semblent bien essentielles 〚28〛.
Par contre, les résultats de Vilette et al. 〚10〛 qui ont montré que des cellules épithéliales de lapin exprimant une PrP bovine sont sensibles aux homogénats de tremblants montrent clairement que des cellules non-neuronales sont capables de répliquer l’agent et qu’il n’est pas nécessaire d’avoir une lignée cellulaire de même origine que la PrP exprimée pour permettre l’infection.
3 Utilisation des cultures cellulaires dans l’étude de la biologie de la PrPC
3.1 Etude du cheminement intracellulaire de la PrPC
Les cultures cellulaires exprimant naturellement ou après transfection des quantités significatives de PrP, ont permis l’identification des grandes étapes du cheminement intracellulaire de la PrP. Des techniques d’immunofluorescence, d’immuno-réplique, d’immuno-précipitation, de marquage métabolique, de marquage par iodination ou biotinylation et de cross-linking ont été utilisées pour réaliser ces études. Ainsi, il a pu être confirmé que la PrP est une glycoprotéine ancrée à la surface cellulaire par une ancre glycero-phosphatidyl-inositol 〚29〛. Comme d’autres molécules glypiées, elle est associée à des domaines membranaires enrichis en cholestérol et en sphingolipides ou DRM pour ‘detergent resistant microdomains’ 〚30, 31〛. Une fois à la surface cellulaire, elle suit un cycle rapide d’endocytose et de recylage au cours duquel elle est clivée dans sa partie centrale 〚32, 33〛.
Malgré les avancées récentes, de nombreuses incertitudes subsistent concernant la biologie cellulaire de cette protéine. Ainsi, il n’y a pas de consensus sur le procédé et la voie d’endocytose de la protéine, sur sa dégradation ou encore sur l’importance que revêt son clivage (pour revue voir 〚33〛).
3.2 Etude de la fonction de la PrP
La détermination de la fonction de la PrP est un sujet d’une importance capitale. L’étude du phénotype des souris transgéniques PrP –/– n’a pas permis de donner de réponse satisfaisante compte tenu des faibles effets produits par l’absence de la protéine 〚19〛. Par contre, en utilisant des cultures cellulaires dérivées d’animaux PrP –/–, des informations plus pertinentes ont été obtenues. Il a ainsi pu être montré que l’expression de la PrP semblait être corrélée à la sensibilité des cultures au stress oxydant, à l’activité d’enzymes telle que la super-oxyde dismutase Cu/Zn ou encore à l’incorporation de cuivre 〚34–36〛. Ceci évoque d’ailleurs la description en culture cellulaire d’une activité ‘anti-oxydante’ de type ‘super-oxyde dismutase’ associée à la PrP 〚34, 37〛. Enfin, le rôle de la PrP dans une cascade de signalisation passant par la src kinase Fyn et impliquant la cavéoline a été mise en évidence dans une lignée neuronale 〚38〛. Ce dernier résultat marque une avancée considérable dans la compréhension du rôle de la PrP. Les prochaines étapes consisteront à déterminer la nature du signal physiologique induisant cette cascade et les conséquences de cette dernière.
3.3 Recherche des partenaires et des récepteurs de la PrP
La recherche de partenaires et de récepteurs de la PrP a été poursuivie in vitro notamment par des approches en double hybride 〚39, 40〛. En culture cellulaire, très peu de molécules ont pu être caractérisées. Cela tient probablement aux difficultés à isoler la PrPC native sous forme soluble pour des études de ‘binding’. D’autre part, l’affinité relative de la PrP aux glycosaminoglycanes 〚41〛 rend difficile la caractérisation à la surface cellulaire de récepteurs de haute affinité 〚42〛. Parmi les molécules qui semblent les plus intéressantes, on peut citer le récepteur à la laminine 〚43〛, le dystroglycanne 〚44〛 et la cavéoline 〚38〛.
4 Utilisation des cultures cellulaires dans l’étude de la biologie de la PrPSc
4.1 Etude de la localisation de la PrPSc
L’obtention de cultures cellulaires infectées a permis d’étudier la localisation de la forme pathologique de la PrPSc. Ces études sont cependant délicates car pour différencier la PrPC de la PrPSc, on utilise les propriétés de résistance à la digestion par la protéinase K de la PrPSc (un traitement qui préserve mal les structures cellulaires). Dans les cultures, la PrPSc a été premièrement retrouvée concentrée dans les lysosomes 〚45〛. Par la suite des études ont montré que la PrPSc était associée au DRM 〚46, 47〛 et présente en quantité non négligeable à la surface cellulaire 〚13〛. L’enjeu de cette localisation cellulaire repose sur la détermination du compartiment cellulaire impliqué dans la conversion.
4.2 Étude de la conversion de la PrP
Disposant de modèles cellulaires dans lesquels la PrPSc était produite, il a été possible de mettre en évidence les facteurs importants dans la conversion de la PrP. Des expériences ont montré que la PrPSc était générée à partir de la PrPC endogène vraissemblablement au cours des phases d’endocytose et de recyclage de la protéine 〚48, 49〛. En ce qui concerne les PrP mutées, il semble par contre qu’elles acquièrent des propriétés anormales dès leur synthèse et leur translocation dans le réticulum endoplasmique 〚12〛. Cela suggère qu’un mécanisme de conversion différent existerait dans les formes génétiques d’EST. Les cultures cellulaires ont enfin permis d’étudier en détail les phénomènes de barrière d’espèces. En transfectant des PrP modifiées dans des cultures infectées il a été ainsi mis en évidence des résidus qui joueraient un rôle fondamental dans le passage d’une espèce à une autre 〚50, 51〛. Les différentes tentatives d’infection de cultures cellulaires ont également révélé l’existence de facteurs autres que la PrP elle même qui conditionneraient la propagation de l’agent 〚28, 52, 53〛. Dans l’avenir, les cultures cellulaires seront sans doute un outil essentiel pour la caractérisation et l’étude de ces facteurs.
4.3 Mécanismes de la neurodégénérescence et PrPSc
Les mécanismes exacts de la neurodégénérescence dans les EST restent indéterminés, d’autant plus que la relation entre PrPSc et neuropathologie a été remise en question à plusieurs reprises 〚54–56〛. Afin de reproduire les mécanismes pathogènes des EST, des expériences de toxicité en culture cellulaire ont été réalisées avec des fragments peptidiques issus de la PrP.
Un peptide correspondant à la zone hydrophobe centrale de la PrP (codon 106–126 de la PrP humaine) s’est révélé toxique sur des neurones primaires en culture 〚57〛. Ce peptide, qui a été ensuite utilisé dans plusieurs modèles in vitro, semble activer les cellules microgliales et astrocytaires et ainsi induire un stress-oxydant 〚58, 59〛. Parmi les hypothèses concernant l’effet de ce peptide nous citerons le fait qu’il semble capable de former des pores en s’intégrant dans les membranes biologiques 〚60〛.
En fait, l’une des premières surprises venue de l’étude des cultures cellulaires infectées par les prions a été l’absence d’un effet cytopathologique net lié à la multiplication de l’agent infectieux 〚23–25〛. Certes les cellules PC12 semblaient avoir un phénotype légèrement altéré 〚61〛 et les cellules GT1–7 être légèrement plus apoptotiques après infection 〚27〛, mais ce n’est qu’avec le travail de Milhavet et al. 〚62〛 qu’il a été clairement démontré que l’infection altérait la réponse cellulaire au stress oxydant. Les cellules infectées présentaient notamment une altération de leur activité SOD ce qui n’est pas sans rappeler les résultats obtenus avec des cultures exprimant des quantités variables de PrP (voir paragraphe 3.2.). La relation entre la multiplication de l’agent et le stress oxydant pourrait concerner la fonction normale de la protéine dans le cadre du métabolisme des ions métalliques ou d’une voie de signalisation cellulaire.
Finalement, une forme trans-membranaire de la protéine PrP, appelée Ctm-PrP, a été identifiée et pourrait être le facteur pathogène des EST 〚55〛. L’existence de ces formes trans-membranaires de la PrP a été décrite initialement dans des expériences de traduction in vitro, puis pour des PrP mutées notamment dans des EST d’origine génétique 〚63〛. Il a été également découvert récemment un nouveau gène en aval de celui de la PrP. Ce gène code une protéine aux caractéristiques très proches de la PrP qui a été appelée Doppel. Cette protéine, sur-exprimée dans certaines lignées de souris PrP –/–, induit une neurodégénérescence des cellules de Purkinje 〚64〛. Les cultures cellulaires permettront de définir avec plus de précision la biologie et le rôle de ces molécules dans les EST 〚65, 66〛.
5 Autres utilisations des cultures cellulaires infectées
5.1 Utilisation à des fin de recherche thérapeutique
Les cultures chroniquement infectées représentent un formidable outil pour cribler des substances susceptibles d’interférer avec la génération de PrPSc . Les premières études réalisées dans les années 90, ont permis de montrer que le rouge Congo, ainsi que des dérivés des glycosaminoglycanes, avaient des activités anti-prions significatives 〚67, 68〛. Depuis lors d’autres molécules ont montré en culture une capacité à diminuer la quantité de PrPSc produite: des porphyrines 〚69〛, l’amphotéricine B 〚47〛, des dérivées du rouge Congo 〚70〛, des agents transfectants 〚71〛, des composés chimiques originaux 〚72〛, et enfin des molécules de PrP hétérologues et des peptides issus de la protéine 〚50, 51, 73〛. Au delà de ces tests thérapeutiques, les mécanismes d’action de ces drogues ont également pu être déterminés grâce aux cultures cellulaires 〚42, 47, 74, 75〛.
5.2 Utilisation dans la détection de l’agent infectieux
Les lignées cellulaires sensibles aux prions représentent dans une certaine mesure une alternative aux modèles animaux dans la détection de l’agent infectieux. Dans certain cas, la sensibilité des cultures aux prions est en effet très importante 〚53, 76〛. Il est envisageable d’utiliser de tels modèles dans des buts de diagnostic cependant ces modèles sont limités à certaines souches 〚10, 28, 52, 53〛 et il n’y a pas pour l’instant de lignées cellulaires sensibles à l’agent de l’ESB.
6 Conclusion
Les modèles en culture cellulaire des EST ont pris ces dernières années une place essentielle dans les recherches sur ces affections. Ils permettent en effet d’aborder de façon rapide et efficace les grandes questions qui restent en suspend concernant les prions. Parmi ces interrogations nous citerons : le rôle de la protéine normale, les étapes et les mécanismes de la conversion pathologique en PrPSc, les paramètres influençant la transmission et la propagation des prions. Du point de vue de la recherche appliquée, les cultures représentent également des outils intéressants pour mettre en évidence de nouvelles molécules anti-prion et pour la détection et la caractérisation des différentes souches de prions.