

1 Introduction
The infectious agent associated to transmissible spongiform encephalopathies (TSE) is mainly, if not entirely, composed of a protein termed PrPSc which stands for scrapie isoform of the cellular prion protein or PrPC 〚1〛. The term ‘prions’ used to designate the infectious agent may be defined as pathogenic isoforms of the host-encoded PrPC protein. Prion propagation strictly depends on the presence of PrPC since prnp knock-out mice are resistant to prion infection 〚2〛. It is now accepted that prions could act as a template for the conversion of newly synthesized PrPC into PrPSc in neuronal cells 〚3〛. However, the mechanisms of conversion accounting for prion replication and accumulation are still unclear, and the molecular and cellular events leading to TSE-associated neurodegeneration remain to be elucidated.
One of the current challenges in the field of TSE is the identification of the physiological role of the cellular prion protein. PrP-deficient mice develop normally, are viable 〚4〛 and have but yielded minor clues as to PrPC function. PrPC is a ubiquitous glycoprotein anchored to the plasma membrane by a glycosylphosphatidylinositol (GPI) moiety. Cell surface PrPC may be present either in its native form or as an N-terminally truncated isoform, both isoforms carrying two, one or no Asn-linked sugar chain 〚5〛. Moreover, the proteolytic cleavage involved in the generation of truncated PrPC produces an amino-terminal peptide, which is secreted in the extracellular space. The attachment of PrPC to the outer leaflet of the plasma membrane is consistent with a role in cell surface signalling, cell adhesion 〚6〛, or ligand binding 〚7〛. Candidate PrPC cellular partners have been identified such as the laminin receptor precursor 〚6〛, members of the dystroglycan complex 〚8〛, the Hsp chaperone 〚9〛, an enigmatic protein identified through complementary hydropathy 〚10〛 as well as PrPC itself 〚11〛. Much interest has been raised by the copper binding-ability of PrPC, although the accurate functional link between copper and the prion protein remains to be established. Up to 4 Cu2+ ions may bind to the histidine-containing octarepeats present in the N-terminal domain of PrPC, with a 5- to 10-μM affinity 〚12〛. Recently, an involvement of PrPC in the regulation of the prepsynaptic copper concentration and of synaptic transmission has been proposed 〚13〛. Besides, copper ions appear to stimulate PrPC endocytosis 〚14〛, and, under oxidative conditions, to favour the cleavage of the protein in the octapeptide region 〚15〛. These overall data raise the question as to the involvement of PrPC in the neuronal response to oxidative stress. Indeed, PrP–/– mouse-derived cerebellar cells show an increased susceptibility to oxidative stress as compared to their wild type counterparts 〚16〛. Brown et al. 〚16〛 have also reported a reduced level of Cu–Zn superoxyde dismutase (SOD) activity in knock-out mice. Actually, the PrPC-induced regulation of Cu–Zn SOD may be an indirect effect resulting from copper uptake from the synaptic cleft into the cytosol 〚13〛.
PrPC is abundantly expressed in neurons, which constitute the major targets of TSE-associated cellular damage. The identification of PrP interacting molecules in neuronal cells therefore constitutes a useful approach to unravel PrPC biological function, as well as the phenotypic elements involved in neuronal susceptibility to TSE agents.
2 A model of neuronal differentiation derived from multipotential cells
In order to isolate committed precursors of the neuroectodermal, mesodermal or endodermal lineage, we have developed an immortalisation strategy based upon the introduction of an adenovirus-SV40 recombinant plasmid (PK4) into multipotential cells 〚17, 18〛. The selected clones behave as somatic stem cells 〚19–21〛. They display a stable and undifferentiated phenotype and express genes that are involved in early embryonic development. Upon induction, they have the capacity to undergo differentiation along distinct pathways of a same lineage. Depending on the inducers, they follow mutually exclusive programs up to the end stages of differentiation.
The 1C11 clone, which derives from the F9 teratocarcinoma cell line 〚17, 20〛, is a bipotential progenitor (figures 1A and 1B) with the properties of a neuronal stem cell 〚22〛. 1C11 precursor cells display an epithelial phenotype, express nestin, a neuroepithelial cell marker, as well as genes involved in early neurogenesis such as Notch 1 to 4 and their ligands Delta and Jagged, Krox 20, Mash-1, msx-1, … 1C11 cells do not differentiate in the absence of induction, even when grown in 10 % fetal calf serum, and lack neuron-associated functions.
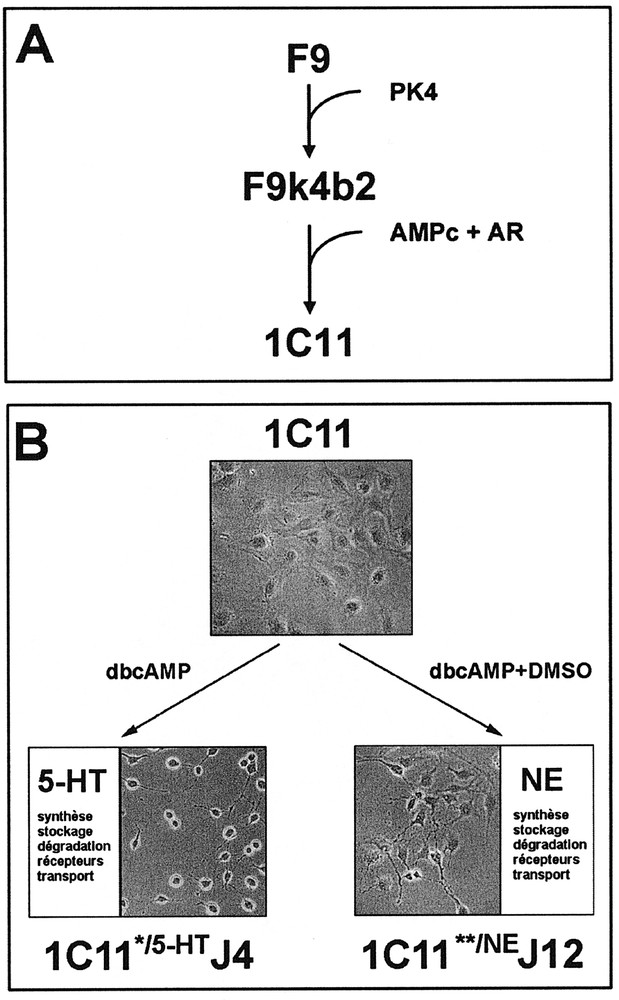
Neuronal differentiation of neuroepithelial 1C11 cells. A. The 1C11 clone has been isolated upon differentiation of F9 embryonal carcinoma cells in vitro. B. Depending on the inducers, 1C11 precursor cells acquire the overall functions of serotonergic (1C11*/5-HT) or noradrenergic (1C11**/NE) neurones.
Within 4 days of treatment with dibutyryl cyclic AMP (dbcAMP) and cyclohexane carboxylic acid (CCA), 1C11 cells convert into 1C11*/5-HT cells expressing a complete serotonergic phenotype. As soon as day 2 after the addition of the inducers, 1C11*/5-HT cells have acquired the ability to synthesize, store and catabolize serotonin (5-HT), and express 2 serotonergic receptors of the 5-HT1B/D and 5-HT2B subtypes. At day 4, a third receptor (5-HT2A) as well as the membranous transporter allowing extracellular serotonin uptake, become functional. Moreover, the receptors selectively induced in the time course of differentiation promote an autoregulation by 5-HT of 1C11*/5-HT cells overall serotonergic functions 〚22〛.
Dimethylsulfoxide (DMSO, 2 %) in combination with dbcAMP and CCA triggers within 12 days the differentiation of 1C11 cells into 1C11**/NE cells, expressing a complete noradrenergic phenotype. 1C11**/NE cells synthesize, store and catabolize catecholamines from day 4 of differentiation, and acquire a functional noradrenergic transporter at day 12. An α1D-adrenoceptor is selectively induced at day 8 of the programme and controls the onset of the noradrenergic transporter 〚22〛.
The two programmes are mutually exclusive. The differentiation events are synchronous within the cell population and concern nearly 100% of the cells (figure 1B). The time sequence of implementation of the neurotransmitter-associated functions is similar to that observed in vivo during neurogenesis. Along either pathway, 1C11*/5-HT and 1C11**/NE cells develop bipolar extensions and express neuronal markers such as neurofilaments, N-CAM, γγ-enolase, synaptophysin and Met-enkephalin.
This differentiation model therefore constitutes a useful tool to investigate the transcriptional and post-transcriptional regulations underlying the onset of neuronal functions and to characterize the involvement of the transduction pathways coupled with the receptors in the expression and/or the maintenance of a bioaminergic phenotype.
3 The 1C11 cell line endogenously expresses the prion protein
The regulation of expression of the prnp gene has been monitored by real-time quantitative PCR analysis (Taqman) 〚23〛. The level of PrPC transcripts present in 1C11 precursor cells is significant as compared to the very low level of expression (< 7 %) measured in multipotential cells, i.e. embryonic stem (ES) cells or the F9 teratocarcinoma cell line, from which the 1C11 clone is derived. These observations are in agreement with those obtained in the time course of embryonic development 〚24〛, and demonstrate that the commitment of multipotential cells to the neuronal lineage is accompanied by a strong increase in the level of PrPC expression.
While no significant variation in the level of PrP trancripts may be monitored during the differentiation of 1C11 cells along the noradrenergic pathway, the entry of 1C11 cells into the serotonergic program is associated with a decrease in the level of PrP mRNA (67 %). This regulation observed at the transcriptional level is paralleled by a modulation of the level of the protein, as assessed by immunoprecipitation experiments 〚23〛. All PrPC isoforms are present in the 1C11 cell line (figure 2).
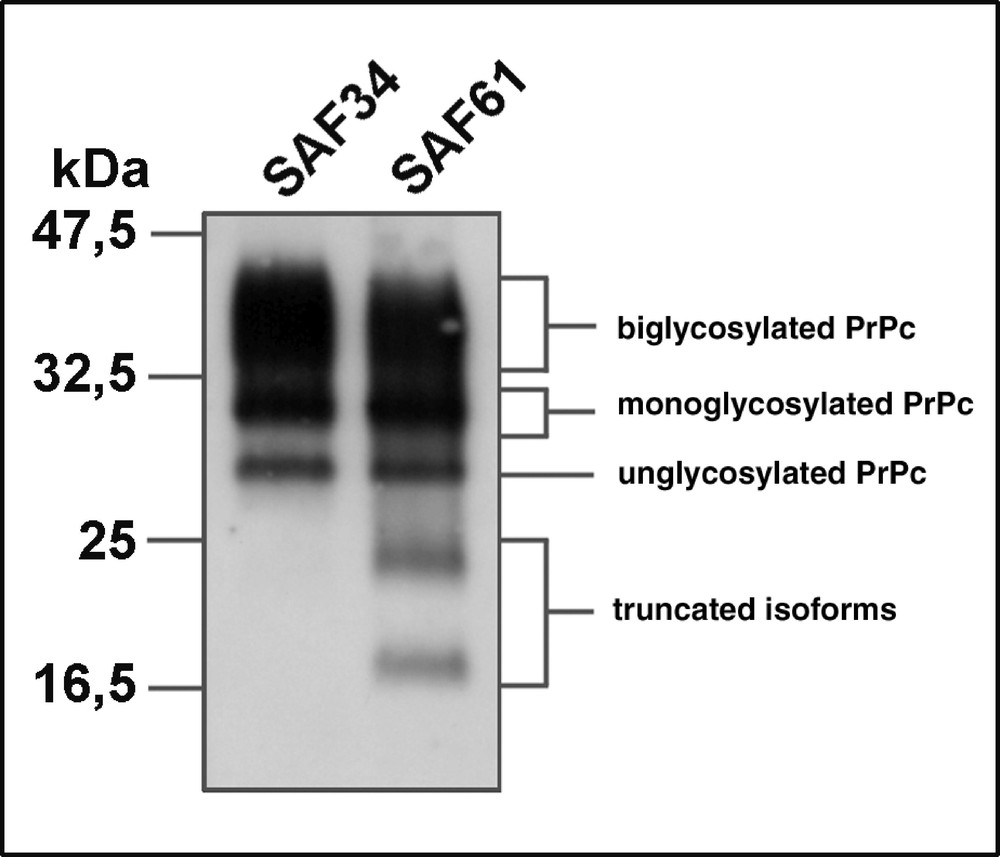
Detection of PrPC isoforms in 1C11*/5-HT serotonergic cells. PrPC immunoprecipitation with monoclonal antibodies directed against the N-terminal (SAF34) or C-terminal (SAF61) domain of PrPC (J.Grassi, CEA Saclay) reveals the presence of PrP isoforms : native, N-terminally truncated, unglycosylated or mono- or bi-glycosylated.
In vivo, the expression of PrPC differs according to brain regions 〚25〛 and the vacuolation pattern observed in TSE-infected brain depends on the agent strain 〚26〛. This suggests a differential expression of PrPC mRNA or isoforms according to the neuronal cell type 〚27〛. However, the accurate distribution of PrP transcripts and protein in relation to the phenotypic specificity of neuronal cells (serotonergic, catecholaminergic, GABAergic, cholinergic, etc.) remains to be characterized. Similarly, besides several studies highlighting a NGF-mediated modulation 〚23, 28–30〛, little is known concerning the regulation of PrPC expression by neuronal factors.
PrPC may be monitored by immunofluorescence at the surface of 1C11 cells, whatever their state of differentiation 〚23, 31〛. Anti-PrP antibodies stain both the cellular bodies and the neuritic varicosities of the serotonergic and noradrenergic cells. The subcellular localization of PrPC in neurones is still imperfectly characterized. In vivo, immunocytochemistry experiments have revealed a strong concentration of the protein in the neuropile 〚32〛, where it may be conveyed through fast axonal transport 〚33, 34〛. A synaptic localization of PrPC in the hamster hippocampus has also been evidenced through electron microscopy 〚35〛. The distribution of PrPC at the surface of the soma and the processes of 1C11*/5-HT and 1C11**/NE cells prompted us to investigate into a putative role of PrPC as a cell-surface receptor.
4 PrPC is coupled with a signal transduction pathway
Our experimental approach to identify a signalling pathway coupled with PrPC lies on antibody-mediated ligation, used as a means to mimic an endogenous signal. PrPC cross-linking does not lead to any p21ras or PLA2 activation, or to any IP3 or NO production within a 30-min time frame. By contrast, PrPC ligation induces the dephosphorylation of Fyn, a member of the SRC tyrosine kinase family. This effect is reproducibly observed with different anti-PrP antibodies. Fyn dephosphorylation correlates with the induction of a Fyn kinase activity. The level of Fyn expression, as measured by Western blot, does not significantly vary during differentiation. However, PrP-mediated Fyn activation is restricted to 1C11*/5-HT or 1C11**/NE differentiated cells, and is not observed in 1C11 precursor cells 〚31〛.
PrPC is a GPI molecule, anchored to the outer leaflet of the plasma membrane, while Fyn is an intracellular kinase. We therefore assumed the involvement of intermediate membrane factor(s) in the coupling of PrPC to Fyn. Co-immunoprecipitation experiments allowed us to identify caveolin as a PrPC cellular partner. The PrPC/caveolin interaction is specific of 1C11*/5-HT and 1C11**/NE cells, although caveolin is also present in 1C11 undifferentiated cells. The PrPC-induced activation of Fyn may be abolished following immunosequestration of caveolin, demonstrating that caveolin indeed is one of the protagonist involved in the signalling pathway coupled with PrPC in these neuronal cells 〚31〛. A colocalization of caveolin together with PrPC may be monitored through immunofluorescence experiments, with a prominent labelling of the neurites varicosities of 1C11*/5-HT and 1C11**/NE cells (figure 3).

Membrane expression of PrPC and caveolin on 1C11*/5-HT serotonergic cells. Double-labeling experiments suggest a colocalization (arrows) of PrPC (B) and caveolin (C) on neurites. (A) Corresponding phase contrast of 1C11*/5-HT cells, 4 days after the addition of the inducers. Scale bar = 25 μM.
5 The coupling of the prion protein to Fyn is dependent upon neuronal differentiation
The kinetics of PrPC-mediated Fyn activation in the time-course of 1C11 differentiation shows that the ability of 1C11 cells to respond to PrPC cross-linking is strictly associated with the acquisition of a complete serotonergic or noradrenergic phenotype 〚31〛. However, the three identified partners of the signalling cascade are present in the 1C11 cell line from the stem cell state. The PrP/caveolin interaction does appear to be a critical event since it is observed as soon as two days after the induction of the cells towards either differentiation program.
The onset of a PrPC/caveolin/Fyn complex, therefore, appears to depend on the implementation of the overall neurotransmitter-related functions. Furthermore, the phenotypic conversion of the cells is most likely accompanied by a maturation of the membrane properties, which may also contribute to the spatial organization of the signalling partners. The onset of neuronal polarity and of fast axonal transport may be critical as well. Indeed, cell fractionation experiments demonstrate that PrPC molecules located on neurites mainly account for Fyn activation 〚31〛.
The identification of a signalling pathway coupled with PrPC defines the prion protein as a cell surface receptor (figure 4). It provides a foundation for uncovering the extracellular signal activating PrP in vivo and for dissecting the intracellular targets downstream of Fyn, which are chemically modified in response to PrP activation.

Signal transduction pathway coupled with the prion protein in neuronal cells. A. PrPC is anchored to the outer leaflet of the plasma membrane and interacts with the transmembrane protein caveolin. Fyn, an intracellular kinase of the Src family is not in its activated state. B. Antibody-mediated ligation of PrPC induces Fyn recruitment, dephosphorylation and activation. The PrPC-caveolin-Fyn signalling complex thus supports the transduction of the signal within the fully dfferentiated neuronal cells. C. Caveolin is one of the protagonists involved in the signalling cascade since its immunosequestration abolishes the PrPC-mediated Fyn activation.
6 Discussion
Due to the paucity of cell lines expressing ex vivo a neuronal phenotype, the cellular biology of PrPC has mainly been investigated using transfected CHO cells, N2a or HaB neuroblastoma cells or PC12 pheochromocytoma cells 〚5, for review〛.
The teratocarcinoma-derived 1C11 inducible cell line constitutes a novel in vitro model to gain insight into the biological function of PrPC in relation to the onset and the control of bioaminergic neuronal functions. The restriction of the PrPC-mediated activation of the Fyn kinase to fully differentiated serotonergic or noradrenergic cells suggests that the implementation of a neuronal phenotype and of neurotransmitter-associated functions is not dependent upon this signalling pathway. By contrast, this PrPC-dependent signal could participate to neuronal homeostasis. This idea is in line with knock-out experiments which argue against a critical role of PrPC in neurogenesis and neuronal differentiation. Besides, an interesting observation is that mice expressing an N-terminally truncated version of PrPC (PrP Δ32-121) suffer from neurodegeneration soon after birth 〚36〛. According to Shmerling, the lesions monitored in these mice would result from a signalling deficiency. Our data obtained with the 1C11 cell line supports the notion that PrPC, or at least some isoforms of the protein, plays a role in the signalling network involved in the maintenance and/or the fine-tuning of neuronal functions.
The PrPC-mediated Fyn activation provides a foundation for identifying the effectors downstream of this kinase, which may control neuronal functions. Fyn is majorly expressed in the brain and appears to be involved in multiple signalling pathways. Some targets of Fyn have been characterized and include: (i) the focal adhesion kinase FAK, a key component, together with paxillin, of focal adhesions, which form a structural link between the extracellular matrix and the cytosqueleton and constitute important sites of signal transduction 〚37〛; (ii) α-synuclein, a pre-synaptic protein associated with vesicular transport and involved in neurotransmitter release at the synapse 〚38〛; (iii) protein kinase C θ (θ PKC ) which has been reported as a Fyn substrate in T cells 〚39〛.
Our observation that the activation of Fyn on the neurites of differentiated cells requires a PrPC–caveolin interaction, suggests that the signalling events arise at the level of sphingolipid- and cholesterol-rich membrane microdomains, i.e. ‘DIG’, also known as major sites of signal transduction. The bioaminergic receptors which autoregulate 1C11*/5-HT and 1C11**/NE cells functions are most likely localized on the neurites. The signalling cascade mediated by PrPC may therefore functionally interact with those coupled with the serotonergic or adrenergic receptors and these crosstalks may in fine control neuronal homeostasis.
Identifying the neuronal response to the PrPC-mediated signal and understanding how PrPSc may interfere with the normal function of PrPC during prion infection hence constitute a great challenge for the next few years. The localization of PrPC in DIG appears necessary for the conversion of PrPC into PrPSc 〚40, 41〛; PrPSc formation indeed is abolished in the presence of drugs such as lovastatin which disrupts DIG structure by decreasing the level of cholesterol 〚42〛. By interacting with PrPC within DIG, PrPSc may hinder its association with cellular partners and thus abrogate the PrPC function. Alternatively, PrPSc may constitutively recruit the signalling pathways normally coupled with PrPC. PrPSc accumulation may also indirectly affect the oxidative state of the cell and thereby modify the balance between PrPC different isoforms 〚43〛 or alter the membrane environment of the signalling complex 〚44〛.
Our recent data indicate that 1C11 cells in the precursor state support prion replication and are able to stably propagate the Chandler, 22L and Fukuoka-1 strains (unpublished data). The 1C11 cell line hence constitutes a new in vitro model to investigate the cellular tropism of the infectious agent. It may also help to unravel the mechanisms related to the toxicity of PrPSc in neurones. Dissecting the molecular and cellular events underlying TSE-associated pathogeny indeed is a priority in order to design therapeutic agents with the capacity to inhibit PrPSc formation while preserving PrPC normal function.
Acknowledgements
We are grateful to J.M. Launay and J.L. Laplanche (Hôpital Lariboisière, 75010 Paris) whose collaborations contributed to this work. We thank J. Grassi (CEA, Saclay) for providing anti-PrP antibodies. This work was supported by the Programme de Recherche sur les ESST et les Prions and by grants from : les CNP assurances, l’Institut Pasteur and le CNRS.