

1 Introduction
Dementia deprives the patient from the ability of valid perception of objects, of understanding their relationship, of comparing them, of keeping full memory of them, resulting in the impossibility of proper reasoning: “La démence prive l’homme de la faculté de percevoir convenablement les objets, d’en saisir les rapports, de les comparer, d’en conserver le souvenir complet ; d’où résulte l’impossibilité de raisonner juste” 〚1〛. This impoverishment of faculties is associated with a series of devastating consequences for the patient and his family. It may lead to disastrous effects on the whole social tissue in ageing countries where the prevalence of dementia rapidly increases. In this paper, we will discuss the definition of dementia, the epidemiology of dementing diseases and their increasing weight in ageing countries. We will consider the regions of the brain usually affected in dementia, and will briefly deal with three paradigmatic dementing disorders: subacute spongiform encephalopathies (prion diseases), Alzheimer’s disease, and vascular and mixed dementia.
2 Definition
The definition of dementia has been much debated. Following the Diagnostic and Statistic Manual of Mental Disorders of the American Psychiatric Association, it is characterised by multiple cognitive deficits severe enough to interfere with social or occupational activities. Impairment in short- and long-term memory is considered mandatory. One at least of the following disturbances of higher cortical functioning is to be associated: aphasia, apraxia, agnosia, trouble of executive functioning (ability to think abstractly and to plan, initiate, sequence, monitor and stop complex behaviour). Behavioural disturbances may be associated 〚2〛. Recognition of dementia may be a difficult task, delirium, depressive episodes, so-called ‘age-associated cognitive decline’ and ‘mild cognitive impairment’ 〚3〛 being the main differential diagnosis. Dementing disorders result from various mechanisms, the causes of which are quite different. This modulates their possible prediction and treatment. In other words, dementia is not a disease, but a complex syndrome due to a variety of causes and mechanisms.
3 Epidemiology of dementing diseases and their increasing weight in ageing countries
In Europe, age-standardised prevalence in 11 cohorts from different countries was 6.4% for all causes of dementia, 4.4% for Alzheimer’s disease, and 1.6% for vascular dementia. The prevalence of dementia increased dramatically with age: 0.8% in the group age 65 to 69 years and 28.5% at age 90 years and older. The corresponding figures for Alzheimer’s disease (53.7% of cases) were 0.6% and 22.2%, and for vascular dementia (15.8% of cases), 0.3% and 5.2%, respectively 〚4〛. The incidence of dementia and Alzheimer’s disease continues to increase with age up to age 85 years, after which rates increase in women but not in men 〚5〛. It should be stressed that these figures must be carefully considered in old and very old people, such as centenarians, in whom the diagnosis of cognitive decline and dementia is particularly difficult 〚6〛. Furthermore, although they are valid for the overall assessment of dementia prevalence, they are disputable when dealing with subtypes: the predictive value of the clinical diagnosis of dementia is still poor, as recently demonstrated by a number of prospective clinico-pathological studies 〚7–10〛. For example, the clinical diagnosis of Alzheimer’s disease is confirmed at pathological examination in 8 to 9 cases on 10. However, if pure Alzheimer’s disease is considered, i.e. when cases with other pathologies combined to Alzheimer’s lesions are excluded, the diagnosis is confirmed in less than half a case only 〚8〛. Associated brain pathologies are very frequent in the elderly, either cognitively normal or demented, and their specific effects on mental status remain often difficult to assess 〚10, 11〛.
Nevertheless, the prevalence of dementia increases steadily in ageing countries 〚12–15〛, and, consequently, the level of institutionalisation 〚16〛. In the French study PAQUID, the mean age of onset of dementia was 82.3 years (figure that may be compared to the mean life expectancy for French men in 2001: 75,2, and women: 82,7 years). The median survival time of the persons with dementia was estimated to be 4.5 years 〚17〛.
4 Topography of lesions causing dementia
As expected from the definition of dementia, the regions that are involved in dementing disorders are principally affected in memory, language, praxias, gnosias and executive dysfunction 〚18, 19〛. We will distinguish schematically the cortex and the subcortex.
In the cortex, three main divisions are recognised: the hippocampus is needed for normal memory acquisitions; the isocortex (also called neocortex) covers most of the brain surface and participates in various specialised networks involved, for example, in language, praxias, gnosias, executive functions; and finally the entorhinal area, limited by the rhinal sulcus, the interface region between hippocampus and isocortex. The isocortex may be subdivided into broad functional categories 〚20〛. The sensory inputs (somesthetic, auditory, or visual) reach primary areas. The main characteristics of the signal are extracted in specialised cortical areas. For example, in the visual mode, colour, movement, and three-dimensional perception are selectively analysed 〚21〛 in so-called association cortices. These are named unimodal because only one type of signal – visual, somesthetic, auditory – is treated in each one. The unimodal association cortical areas are connected with large multimodal areas, located mainly in the prefrontal and in the parieto-temporal regions. The information is sent from these areas onto the entorhinal area, which receives signals that have been processed through the various association cortices. This set of cortico-cortical connections from the primary to the entorhinal areas (‘feed-forward’) is paralleled by connections that join the multimodal areas to the primary cortex (‘feed-backward’) (see 〚22〛). These cortico-cortical connections are thought to play a major role in memory processes by providing the necessary relays between the hippocampus and the isocortex.
The subcortex comprises subcortical nuclei and the white matter consisting of myelinated axons connecting the previous structures and the different cortical areas. The normal functioning of the cortex depends not only on its own integrity but also on various subcortical mechanisms, controlling: (1) the influx of afferences bringing information from sensory organs or other cortical areas; (2) the targeting of efferences from other parts of the cortex and the subcortical nuclei; (3) the adequate supply in neuromodulators regulating important functions as attention, arousal, wakefulness. The main projections to the cortex are issued from the thalamus. Non-specific thalamic nuclei project diffusely to the cortex. Among the specific nuclei, reciprocally connected to definite areas of the cortex, the pulvinar is connected to the parieto-temporo-occipital associative cortex, and the dorso-medial nucleus is connected to the prefrontal cortex 〚23〛. In addition, at least five cortico-striatal and cortico-striato-pallidal transthalamic circuits organised into parallel rings have been recognised 〚24, 25〛. Other examples of significant subcortical nuclei are provided by the large population of neurones the perikarya of which are situated in the basal forebrain (including the basal nucleus of Meynert, the medial septal nuclei, the nucleus of the diagonal band of Broca). They provide diffuse cholinergic projections to the hippocampus and the entire isocortex. Similar projections arise from the tegmental ventral area, locus ceruleus and the raphe nuclei, and provide dopamine, noradrenaline and serotonine innervation to the cortex, respectively 〚26〛.
5 Mechanism of dementia
5.1 Cortical dementia
The cortical lesions leading to dementia may be diffuse or focal. Destruction of large cortical areas, occurring in multiple infarcts, can thus induce dementia by simple addition of cognitive deficits. It has been demonstrated by morphometric methods that the volume of damaged tissue, always greater than 50 ml in the studied series, was correlated with the severity of cognitive impairment 〚27〛. The minimal volume of brain tissue (cortical and subcortical) that must be destroyed has been debated. One hundred millilitres and more were generally associated with dementia in the pioneer study of Tomlinson et al. 〚28〛, but these authors found infarcts larger than 50 ml in their control group and smaller than 100 ml in patients with purely vascular dementia. Tomlinson et al. concluded that the threshold depends on the site of the lesions in the brain and proposed the concept of strategic sites. Indeed, small strategic infarcts may produce a profound and diffuse disturbance of cognition 〚29〛. Bilateral infarcts are usually responsible but, when unilateral, the left side is more commonly affected 〚30〛. Among the unilateral cortical infarcts that may cause symptoms of dementia, those involving the angular gyrus (which induce fluent aphasia, alexia with agraphia, memory disturbance, spatial disorientation and constructional apraxia) have been identified. The clinical consequences of the large infarcts due to occlusion of the left middle cerebral artery, causing aphasia, have been discussed for almost a century. Pierre Marie, at the onset of the 20th century, thought that aphasia was associated with a “deep reduction of intelligence”, while Dejerine maintained at the same time that it was purely a language deficit (for review of the ‘Controversy on Aphasia’, see 〚31〛). More recently, some clinical criteria still prefer to dodge the issue: “Patients with middle cerebral artery occlusions presenting with severe aphasia are excluded from the vascular dementia group since appropriate evaluation is difficult” 〚29〛.
5.2 Subcortical dementia
1. Dementia, or similar symptoms, is sometimes related to indirect disturbances of various areas of the cerebral cortex, either by disorders of cortical afferences or efferences, or by an inadequate supply in neuromodulators 〚32〛. This describes a cluster of symptoms, which include forgetfulness, slowness of thought (‘bradyphrenia’), emotional or personality changes and impaired ability to manipulate acquired knowledge, whereas aphasia, apraxia and agnosia (‘cortical deficits’) are noticeably absent 〚33〛. Such an association of symptoms frequently occurs when the lesions affect predominantly the striatum, thalamus or subcortical white matter, and is reminiscent of that observed in lesions of the prefrontal cortex (the term subcortico-frontal dementia is sometimes used). It may be added that the auto-activation deficit, related to basal ganglia dysfunction, has to be differentiated from dementia 〚34〛.
2. The best examples of subcortical dementia are provided by arterial occlusion. Infarcts of the paramedian (thalamo-perforating) territory of the posterior cerebral artery may cause a bilateral butterfly-shaped lesion of the intralaminar nuclei of the thalamus, often involving the mamillo-thalamic tract, and presenting with dementia (‘thalamic dementia’) 〚35〛. Dementia or dementia-like syndromes have also been observed in other subcortical infarcts, involving most noticeably the head of caudate nuclei 〚36〛 and the genu of internal capsula 〚37〛. This last example demonstrates that the disconnection induced by necrosis of a white matter tract may cause a deep, if transitory, alteration of cortical functioning.
3. That a dementia may have a subcortical origin was first assumed in degenerative disorders such as Progressive Supranuclear Palsy, Alzheimer’s, and Parkinson’s diseases 〚38〛. The distribution of lesions is not as clearly settled in degenerative cases as in vascular pathology, however. A purely subcortical or cortical origin of dementia is questionable in most degenerative diseases. Even if rarer than in some basal ganglia and brain stem structures, neurofibrillary tangles have been recognised in the cortex of Progressive Supranuclear Palsy 〚39, 40〛 and could thus cause directly, or contribute to, the deficit. In Alzheimer’s disease, the cholinergic hypothesis holds that neuronal loss in the nucleus basalis of Meynert, which innervates the cortex with acetylcholine, plays an important role in the cognitive deficit 〚41〛. Deprivation of the cortex in that neurotransmitter is thought responsible for at least part of the symptoms but cortical lesions (neurofibrillary tangles and senile plaques) play also a role. The intellectual deficit sometimes present in Parkinson’s disease has been, at least partly, attributed to a subcortical cause, the destruction of the internal part of the substantia nigra disconnecting the cerebral cortex from its meso-cortical afferents 〚42〛. However, sensitive techniques developed recently, such as Tubulin Associated Unit and alphasynuclein immunohistochemistry or special silver stains, have shown that the lesions of Parkinson disease are more largely distributed than previously thought, the cerebral cortex and the amygdaloid body, involved in cognitive and emotional processing, being affected 〚43〛. For these reasons, Parkinson disease cannot be considered any more as a purely subcortical disorder. Even if neither the subcortex nor the cortex is affected in isolation in these disorders, however, associated lesions may contribute to, and modulate, the cognitive deficit.
4. White matter lesions have been incriminated for long in the pathogenesis of cognitive deficits: some cases of multiple sclerosis – particularly when the corpus callosum is involved – present with a dementia 〚44〛. Dementia is also known in diffuse white matter lesions seen in leukodystrophy or in Binswanger’s encephalopathy associated with arterial hypertension 〚45〛. It is also one of the symptoms of CADASIL (cerebral autosomal dominant angiopathy with subcortical infarcts and leukoencephalopathy) 〚46, 47〛 where the cognitive deficit has been considered to be of purely subcortical origin 〚48〛.
5. Many structures, often associated, are thus involved in the various disorders leading to dementia in human beings. Simple experimental models are needed. Although mandatory, however, they usually fail to explain easily every refinement of human mind, and the complexity of its pathology. Studies in human should always be kept in mind when assessing the validity of a simple hypothesis in a human model.
6 Some paradigmatic dementing disorders
6.1 Subacute spongiform encephalopathies (prion diseases)
1. Subacute spongiform encephalopathies are rare disorders characterised by widespread neuronal vacuolation (Fig. 1A) and death associated with the presence of a misconformed molecule, called either PrPsc (Prion protein of scrapie) or PrPres (Prion protein resistant to proteinase K). In human, they can occur as sporadic, genetic or infectious diseases. Increase in the number of β-pleated sheaths in the PrP molecule leads to an exceptional resistance to denaturing agents. It is associated with an increase in the affinity of the molecule for special stains, such as Congo red, that help to define ‘amyloid deposits’. It may, finally, be the support of infectivity according to the prion theory 〚49〛. In some cases, large amounts of PrPres accumulate in the extracellular space, forming amyloid aggregates of various shapes, named plaques (Fig. 1B). In human diseases, such as Creutzfeldt–Jakob disease, the lesions usually involve most brain structures implied in mental function: various isocortical areas and the entorhinal cortex, basal ganglia, especially the striatum and the thalamus 〚50, 51〛. The distribution of changes, which also involve widely the hippocampal formation in some variants, is largely modulated by a polymorphism on codon 129 of the PRNP, the gene coding for PrP 〚52〛, and this leads to different clinico-pathological variants 〚53〛. The various glycosylation levels of PrPres, which modulate in part its migration pattern in Western blotting and have led to the description of different types of PrPres, may also be involved in the phenotypic expression of Creutzfeldt–Jakob disease 〚54〛. The pathogenic mechanisms are likely more complex: for instance, it has been recently shown that two or more different types of PrPres could occur in the same brain 〚55〛.
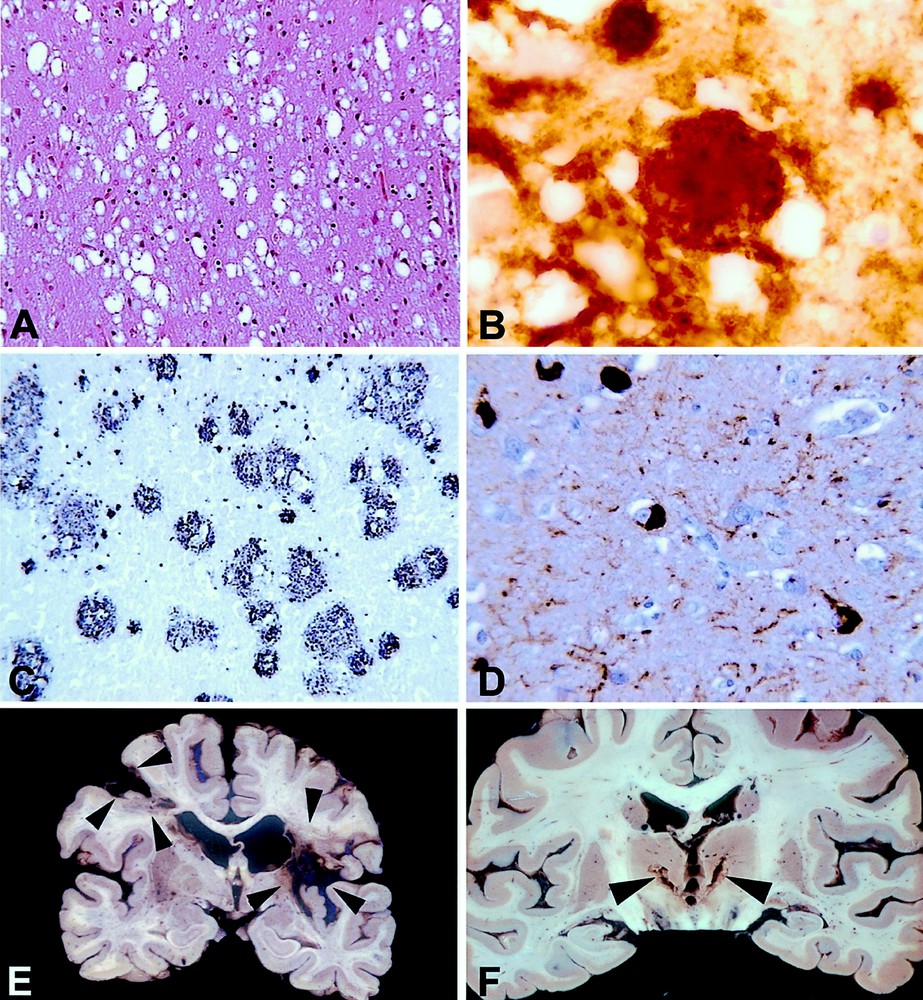
A and B: Creutzfeldt–Jakob disease. Microscopic lesions in the cerebral cortex. A: Sporadic Creutzfeldt–Jakob disease. Numerous cavities in the neuropile indicate a spongiform change. This characteristic lesion is due to the development of vacuoles in the neuronal cytoplasm, especially in the processes. Paraffin embedding, hematoxylin and eosin stain, Objective × 20. B: New variant of Creutzfeldt–Jakob disease. A florid plaque is shown by PrP immunohistochemistry. A large rounded PrP deposit labelled by the antibodies is surrounded by vacuoles of spongiform change. Paraffin embedding, 12F10 anti PrP monoclonal antibody, generous gift of Dr J. Grassi, CEA, peroxidase-antiperoxidase (AAP) with diaminobenzidine (DAB) as a chromogen. Objective × 100. C and D: Alzheimer’s disease; microscopic lesions in the cerebral cortex. C: A large variety of plaques are demonstrated by Aβ immunohistochemistry. Paraffin embedding, 6F/D3 anti Aβ monoclonal antibody (Dako®), PAP and nickel-enhanced DAB. Objective × 20. D: Neurofibrillary tangles and neuropil threads are stained brown by anti-tau immunohistochemistry. Five neurones containing abnormally phosphorylated tau are seen. The thin and tortuous fibres are called ‘neuropile threads’. Both lesions consist of abnormal filaments made of hyperphosphorylated tau protein. Paraffin embedding. Rabbit polyclonal anti tau antibody (Dako®), PAP and DAB. Objective × 40. E and F: Macroscopic examples of brain sections in vascular dementia. E: In this case, two large infarcts are present in both hemispheres (arrow-heads). These massive areas of brain destruction explain the cognitive deficit. F: In another case, two small nearly symmetrical infarcts are seen in the thalamus (paramedian thalamic territories). This is an example of isolated lesions of strategic areas leading to dementia.
2. The main brain structures implied in cognitive deficits responsible for dementia are involved in prion diseases. Although quite rare, these diseases are of considerable interest, however, for two main reasons.
- • The cause and mechanism of the neuronal dysfunction and death remains poorly understood: unconventional infectious agents, or prions, that accumulate in the brain remain biological enigmas. That the PrP protein is one of the main actors in the development of the disease is fully accepted. If we must give only one argument for this, it is brought by the studies on knock out and transgenic mice: Prion diseases do not develop in PrP knock out mice. Furthermore, when a brain graft from a normal mouse into the brain of a PrP knock out mouse is inoculated, the histological alterations due to prions do not cross the border of the graft and spare the brain of the knock out mouse. When transgenic mice acquire the hamster PrP gene, they become susceptible to inoculation by infected brain from hamster 〚56〛. That the PrP protein is the only one to act in Prion disease is more controversial. As long as the transconformation of synthetic PrP ‘in vitro’ does not lead to an infectious molecule, this debate will be open.
- • Whatever the mechanism of their development, prions cause public health concern. No drug has been demonstrated so far to be useful in human prion diseases. Whether PrP storage and the spongiform lesion of neuronal processes are reversible will govern the chances of curative treatments. Although the prevalence of the new variant of Creutzfeldt–Jakob disease is extremely low (up to now, six cases have been recognised in France, five of which only being fully confirmed by post-mortem brain study), preventing the development of this disease is mandatory: In contrast with classical Creutzfeldt–Jakob disease, not only the brain, but the lymphoid and other tissues are infectious, and this may lead to iatrogenous infection 〚57, 58〛. To prevent the few primary cases of the present epidemic spreading to secondary cases is a major challenge. Such a prevention, which commands reinforced epidemiological surveillance, would hinder the development of an endemic state difficult to control because of the lack of peripheral marker of the disease, the exceptional duration of the incubation time, and the extraordinary resistance of the agent. It may be recalled that he only human prion disease ever eradicated has been ‘kuru’: preventing funeral cannibalism led to the disappearance of this devastating disease in the Fore people of New Guinea 〚59〛.
6.2 Alzheimer’s disease
Alzheimer’s dementia, the prevalence of which is the highest among the dementing disorders observed in industrialised countries, raises a number of different questions, some of which govern the possibilities of treatment. The cause of the disease, the reason for the selective distribution and development of the lesions, and their relationship with the so-called physiological ageing remain poorly understood.
1. The abnormal metabolisms of Aβ peptide and tau protein (Figs. 1C and D), which are under elucidation by cell and molecular biology methods 〚60–62〛, follow neuro-anatomical pathways. This is especially valid for tau-related pathology 〚63, 64〛. The spatiotemporal sequence of lesions complies with a hierarchical model following the brain systems involved in memory processing: the entorhinal cortex, then the hippocampus, and lastly the isocortical associative areas being successively affected 〚65〛. This led to propose a neuropathological staging of Alzheimer-related changes 〚66〛, which correlates with the pre-mortem intellectual status, assessed by psychometry 〚67〛. Tau-associated lesions occur very early in some persons 〚68〛, and tend to be constant in centenarians 〚69〛. The frequency of the stages of Alzheimer-related changes in different age categories 〚70〛 is linked to the age-related prevalence of dementia 〚71〛.
2. Which is the first alteration, Aβ peptide accumulation or tau protein dysphosphorylation? No clear answer has been brought up to now. Some authors emphasise the cases of Alzheimer’s disease associated with genetic disorders increasing the production and/or deposition of Aβ peptide in the brain 〚72〛. Others point to the early Tubulin Associated Unit-associated lesions seen in the entorhinal cortex in young adults 〚70〛. A neuropathological observation brings some information on this topic. In a small piece of isocortex disconnected 27 years earlier by the neurosurgical removal of a benign tumour, the density of lesions associated to Tubulin Associated Unit dysphosphorylation was close to zero, whereas the diffuse deposits of Aβ were abundant 〚73〛. This indicates that neuritic and Aβ pathologies may be dissociated in the same individual and suggest that the tau-associated alterations mainly involve cortico-cortical fibres coursing tangentially in the cortical ribbon.
3. Whether Alzheimer’s disease is the mere consequence of brain ageing, and thus an ineluctable age-related process, or a genuine pathological disorder modulated by a number of risk factors, age being the most significant, is still debated. The answer heavily relies on the understanding of normality in ageing 〚74, 75〛. Whatever the chosen concept of normality, several data support the second hypothesis, however: In some families, four different genetic disorders increasing the deposition and/or deposition of Aβ peptide in the brain are associated with diseases that occur in young patients and are very similar to sporadic Alzheimer’s disease 〚72〛. On the other hand, in addition to age, a number of risk factors for Alzheimer’s disease (which may favour its development, such as trisomy 21, ApoE4 genotype and head trauma, or may prevent it, such as the use of non steroidal anti-inflammatory drugs 〚76〛) have been described 〚64〛. Lastly, in centenarians, Alzheimer’s disease can be differentiated from ‘normal ageing’ 〚69〛. Alzheimer’s disease may thus be viewed as a complex syndrome linked to various risk factors, either genetic or linked to environment, which modulate the speed of accumulation of brain lesions associated with Aβ peptide accumulation or Tubulin Associated Unit dysphosphorylation. Some of these risk factors could be accessible to prevention or to curative therapy.
4. The main developments in present knowledge of the neuropathology of Alzheimer’s disease have been brought by prospective clinico-pathological studies following the pioneer work of Tomlinson 〚28〛. These include, in France, the Charles Foix longitudinal study 〚65〛. The duration of such studies, their cost and the declining rate of autopsy have reduced their number, and deprive us of precious data that might have opened new research fields.
6.3 Vascular and mixed dementia
1. Vascular dementia, the prevalence of which is debated because non-controversial diagnostic criteria, both clinical and neuropathological, are lacking, introduces another paradigm: In most instances, the cause of the disease is recognised, leading to possible prevention. Vascular dementia provides, however the unique opportunity for clinico-pathological correlation, provided that the lesions are pure, and that the patient has been prospectively examined. As detailed in the chapter ‘Mechanism of dementia’, infarcts either destroying large volumes of brain tissue or involving the territories of some cerebral arteries, i.e. anterior or posterior cerebral arteries, are more frequent in dementia related to stroke (Fig. 1E and F). It has been recently demonstrated by a prospective clinico-pathological study followed by a morphometric analysis that this could be explained by the infarcts destroying selective strategic areas: these included heteromodal associative areas, the hippocampus and related structures of the limbic system 〚77〛.
2. Unexpectedly, a series of recent reports have demonstrated that brain lesions due to vascular mechanism were often associated with the lesions characteristic of Alzheimer’s disease, and that both were frequent in non-demented persons enrolled in prospective and epidemiological population studies 〚7–11〛. This renewed the concept of ‘mixed dementia’ 〚78〛, which has been very much debated. There was even doubt as to whether mixed dementia actually exists as a clinical entity and some authors have recommended the revaluation and more detailed redefinition of this term 〚79, 80〛. A recent prospective clinico-pathological study showed that patients with few Alzheimer-type changes, but with some additional vascular lesions, were as demented as those with more Alzheimer-type changes but with no vascular lesions. This provides support for the validity of the mixed dementia concept and suggests that the pathological processes involved in AD and vascular dementia are synergistic and cumulative 〚27〛. Such observations may be due to interrelated mechanisms such as amyloid angiopathy, present in Alzheimer’s disease, and which can cause cerebral infarcts and haemorrhages, and thus induce a vascular dementia 〚81〛, or to the involvement of some common pathological pathways, such as cholesterol metabolism, which may be altered in both Alzheimer’s disease and vascular dementia 〚82〛. These facts must be kept in mind when interpreting the data, but trivial reasons (such as early recognition of incipiens Alzheimer’s disease when a vascular lesion occurs in the brain) may also be responsible for the observed association between Alzheimer pathology and vascular brain lesions. Indeed, the recognition of mixed dementia (where degenerative lesions – mainly of the Alzheimer type – and cerebral pathologies of vascular origin co-exist in the same patient) appears more difficult than the diagnosis of pure mechanisms. A case diagnosed as ‘pure Alzheimer’s disease’ could actually be a mixed dementia. This diagnostic error would falsely assign to Alzheimer’s disease the risk factors of vascular diseases.
3. Increasing the diagnostic accuracy of Alzheimer’s disease, and generally of dementia, is a difficult goal, because non-invasive markers are lacking. With a few exceptions, cerebral biopsy, i.e. sampling of brain in the living person, cannot be considered as an ethical method for diagnosis or research in the field of dementia 〚83〛. In this regard, autopsy, i.e. post-mortem examination, is a far more ethical procedure 〚84, 85〛, and provides together full diagnostic means and large opportunities for post-genomic research 〚86〛. Further prospective studies should include as much as possible the post-mortem certification of diagnosis, and the results must be carefully discussed 〚87〛.
7 Conclusion
The neuropathology of dementia provides complex data, and these need to be coped with simpler models. In turn, the intricate lesions of human brain leading to dementia should always be kept in mind when assessing the validity of unifying concepts. Distinguishing and accurately diagnosing the various types of dementia is essential for understanding their mechanism and for developing efficient therapeutic strategies, preventive and curative. For such objectives, the study of human brain remains mandatory. Until non-invasive markers and additional models are available, brain tissue has to be studied. Ethical reasons dismiss the use of cerebral biopsy and favour the promotion of autopsy.
Acknowledgements
We wish to thank the members of the ‘Laboratoire de neuropathologie Raymond-Escourolle', and INSERM U 106 and 360 for constant help.
Version abrégée
La prévalence des démences augmente très rapidement au cours du vieillissement, ce qui grève de la crainte d’un handicap sévère les espoirs portés par la prolongation de l’espérance de vie. La définition des démences, les principales données sur leur épidémiologie, les structures cérébrales impliquées sont discutées. Les démences relèvent de causes et de mécanismes très divers, qui influent sur les possibilités prédictives et thérapeutiques. Trois d’entre elles sont évoquées schématiquement
- • Les affections à agents transmissibles non conventionnels (ou « prions ») fournissent un exemple dans lequel la dégénérescence et la mort neuronale, parfois massives, affectant la majorité des structures cérébrales impliquées dans les fonctions supérieures, expliquent aisément la démence. Il reste à comprendre le mécanisme de ces maladies et à établir la réversibilité des lésions les plus caractéristiques, le dépôt de protéine PrP amyloïde et infectieuse et la spongiose neuronale, pour évaluer les chances de traitements curateurs. Malgré leur grande rareté, les maladies humaines à prions constituent un défi pour la santé publique, en raison, notamment, des risques de contamination iatrogène que fait courir la nouvelle variante de la maladie de Creutzfeldt–Jakob. Leur surveillance épidémiologique, qui repose sur l’autopsie, doit être renforcée.
- • La maladie d’Alzheimer, la plus fréquente des démences, soulève plusieurs questions différentes : sa cause, encore inexpliquée, la raison de la topographie sélective et du mode d’extension stéréotypé des lésions qui la caractérisent et, enfin, ses rapports avec le vieillissement cérébral, considéré comme physiologique. Dues au métabolisme anormal du peptide Aβ et de la protéine tau, les lésions présentent une topographie cérébrale sélective. L’extension topographique de la phosphorylation anormale de la protéine tau se fait selon une progression hiérarchique stricte inexpliquée, qui paraît suivre les circuits impliqués dans la mémoire. Cette modalité de propagation est d’autant plus importante à comprendre que ces lésions sont très précoces chez certaines personnes et deviennent constantes chez les centenaires. Une partie d’entre elles paraît relever de la simple sénescence, la pente de la courbe qui décrit leur production au cours du temps étant modulée par des facteurs de risque génétiques et épigénétiques.
Les démences vasculaires, dont la fréquence reste discutée, offrent un autre paradigme à étudier : leur cause est, dans la majorité des cas, connue. Elles autorisent, lorsqu’elles sont pures, des corrélations clinico-pathologiques précises, permettant l’étude des structures dont la lésion est responsable de la démence. Le volume de tissu cérébral détruit et l’atteinte de régions stratégiques jouent des rôles intriqués. D’autres part, des lésions d’origine vasculaire sont fréquemment associées à celles de la maladie d’Alzheimer. Elles fournissent un exemple d’incertitudes importantes persistant dans le diagnostic clinique des démences, ce qui conduit à des difficultés pour l’interprétation de certaines données épidémiologiques.
L’intérêt, pour la confirmation diagnostique et la recherche, de l’étude du cerveau humain dans ces trois démences est souligné. En l’absence de marqueur fiable et facile à utiliser des maladies responsables de la grande majorité des démences, l’examen du tissu cérébral reste indispensable pour obtenir la certitude diagnostique et permettre la recherche génomique et post-génomique. L’autopsie est, dans ces cas, bien plus éthique que ne l’est la biopsie cérébrale pratiquée sur le malade vivant.