

Discoveries of cerebral synthesis and metabolism of “neurosteroids” [1] have led to the study of new active compounds usable in presently poorly treated human pathological conditions.
We summarize here two quite different examples. Firstly, we deal with a synthetic derivative of pregnenolone (PREG), a steroid metabolically formed from cholesterol, 3β-methoxy-pregnenolone ([MAP4343], Fig. 1), which is effective for treating the damaging consequences of traumatisms of the central nervous system and abnormal cerebral function associated with depressive states; its original therapeutical activities have been already very much studied [2,3]. Secondly, we report on a protein (FKBP52), discovered as a component of hetero-oligomeric steroid receptors and cloned 22 years ago in my laboratory [4]; its physical and functional interaction with the pathogenic tau protein observed in the Alzheimer's disease and other senile dementias is now well established [5–7].
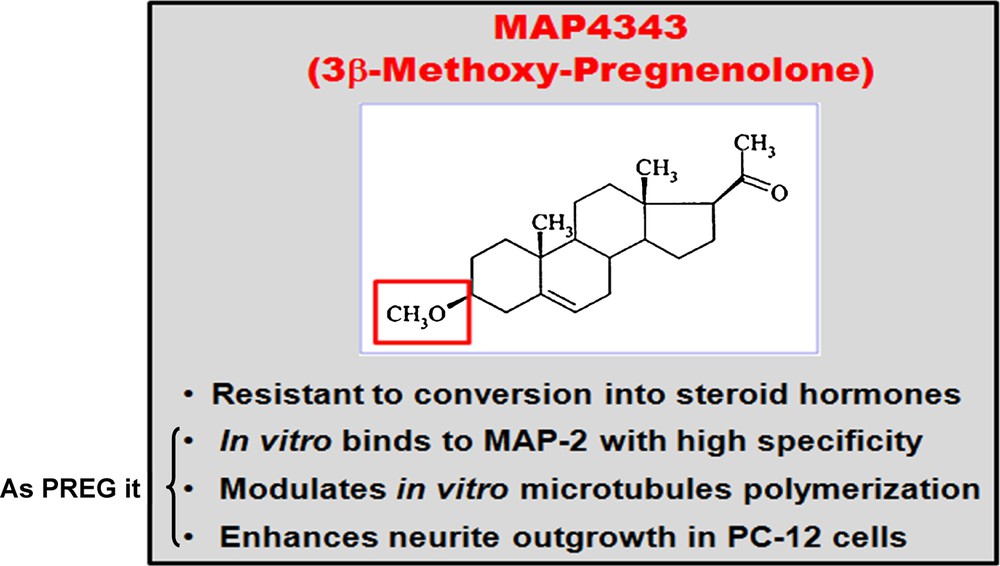
(Color online). MAP4343, a pregnenolone derivative.
In both cases, the effects of MAP4343 and FKBP52 have nothing in common with classical hormonal function of steroids, and the involved mechanisms are different from everything so far described in the hormonal field. Studies of microtubules, cellular structures that ensure vital structure and function in neurones are at the heart of the main pathophysiological phenomena associated with lesions and treatments reported in this short paper.
1 Treatment of depression: a most frequent neurobiological pathology
Depressive disorders are complex pathological states resulting from an unidentified gene mechanism–environment interaction. Their burden is currently the second highest in developed countries (cardiovascular disorders rank first). According to the World Health Organization, it permanently affects more than 120 million people over the world. Approximately 10% of men and 20% of women over their lifetime suffer from a major depressive episode. Depressed individuals experience high rates of co-morbidities and mortality during cardiovascular dysfunctions. Sixty to 70% of acutely depressed patients experience suicidal ideation, and 10–15% commit suicide.
1.1 Conventional anti-depressant drugs: limitations
Post-mortem human and animal studies show that long-term administration of conventional anti-depressants sometimes help to recover the neuronal structure alterations observed in depression. In contrast, short-term administration of many currently used anti-depressant drugs exerts aversive effects on neuronal functions.
Anti-depressant drugs require a 2–4 weeks administration to reach therapeutical efficacy in depressed patients. Anti-depressants such as Selective Serotonin Reuptake Inhibitors (SSRIs) often cause adverse “mood” effects during the first weeks of treatment, including insomnia, anxiety, and cognitive defect. About 30% of patients are non-responsive to SSRIs, and the Food and Drug Administration requires Black Box warnings on all SSRIs, which state that they double suicidal rates (from 2 in 1000 to 4 in 1000) of children and adolescents.
Recent observations have described abnormalities of neuronal microtubules in patients suffering from depressive states [8]. Microtubules form an intracellular network of micro-tubes, crucial to cell structure and function. They are made of tubulin α and ß units, whose assembly and activity are influenced by Microtubule-Associated Proteins (MAPs).
Our strategy involves rescuing therapeutical damages by improving pharmacologically the structure/activity of microtubules. We have identified a way to target and restore altered neuronal microtubules: MAP2 acts as a specific functional receptor of PREG in the CNS [9]; PREG and MAP4343 bind to MAP2 and promote microtubule polymerisation of tubulin and neurite outgrowth.
1.2 The therapeutic properties of 3B-methoxy-pregnenolone (MAP4343) tested in 3 animal models of depressive disorders
In the social isolation model (rats), stress is induced by isolating the animals during six weeks [3]; in the Tree Shrews psycho-social stress model, stress is induced by repeatedly exposing some males to dominant other males [10]; in the Wistar Kyoto (WKY) rat model, the animals suffer from spontaneous/endogenous severe depressive disorders [11]. In all cases, MAP4343 fills the therapeutic gap, and this was demonstrated in a competitive innovative program granted by the European Union Eureka/Eurostars “Depression and Steroids” grant E!5291 (“DEPSTER” project). In these three different animal models studied by our group, MAP4343 promoted “coping” (= survival) behaviour, decreased anxiety, restored memory, and cognitive function, improved sleep efficacy, displaying an immediate and persistent activity (faster and better than in systematic comparison of Prozac action), no side-effect and no sign of drug-abuse. The activity was correlated with some rapid modifications of tubulin markers in the brain.
MAP4343 is a pregnenolone derivative resistant to metabolic conversion into steroid hormones, contrary to PREG itself. It does not have any hormonal/anti-hormonal activity. In vitro, it binds to MAP-2 with high specificity, modulates microtubules’ polymerisation, and enhances neurite outgrowth in PC-12 cells. The phase-I study has been successful in human beings, indicating the safety of the drug (unpublished). MAP4343 favours an effect of assembly of microtubules. Interestingly, in the endogenously depressed WKY model, rats are proved resistant to treatment with SSRIs (Prozac and Escitalopram), fluoxetine even degrades coping behaviour, but MAP4343 restores it.
Phase-II studies of MAP4343 will be incessantly conducted in Paris according to an add-on program at the Hospitals Saint-Anne (Profs. R. Gaillard and M.-O. Krebs) and La Salpêtrière (Prof. P. Fossati).
2 Senile dementias: Alzheimer's disease and other tauopathies
Since Dr. Alois Alzheimer described them in 1907 [12], characteristic biochemical abnormalities have been observed in the largest number of senile dementias called after the Austrian psychiatrist. It is believed that there will be more than 50 million cases a year defined as Alzheimer's disease during the second part of the 21st century. Inter-neuronal amyloid plaques invade the brain from the hippocampus, and intra-neuronal tangles, principally made of tau protein in neurones, altered mostly by hyperphosphorylation and limited truncation [13], are observed. Besides Alzheimer's disease, other dementias, named Pick disease, Progressive Supranuclear Palsy, Frontotemporal Dementia, and Parkinsonism, linked to chromosome 17, etc., also are “tauopathies”, but do not exhibit plaque formation. They differ somewhat biochemically and clinically one from the others, but all are severe diseases leading to neuronal alterations till destruction and synapses dysfunction with consequently loss of memory, defective gnosis, and decrease of consciousness for several years. Recently, pathological forms of the Tau protein have been recognized as very important if not central in terms of molecular responsibility for bio-clinical evolution of these diseases, and prion-like protein displacement are now considered as involved in the evolution of pathological lesions. All tauopathies are clinically established after some years (10–20 years and more) of biological development. Presently, there is no active treatment, but it is logically believed that when we shall become able to early interrupt the pathological process, we will have a better chance to cure patients.
We have described in 2010 [5] the physical interaction of tau protein with FKBP52, a cytoplasmic protein that we discovered as a component of the hetero-oligomeric structure of steroid hormone receptors that we isolated more than 15 years ago [4]. FKBP52 is decreased in Alzheimer brains [6] and it is partly localized and metabolized in lysosomes [14]. FKBP52 is an immunophilin whose structure includes an N-terminal peptidyl-prolyl isomerase site able to bind to immuno-suppressant drugs such as FK506, which inhibits its enzymatic activity; however, FKBP52, whatever it binds to in terms of ligands, including antidepressants, does not promote anti-immunological activity as other members of the class of immunophilins. When it binds to tau protein, FKBP52 can stimulate an anti-tau activity (Fig. 2), including when tau protein is pathologically (including with tau P301L mutation) involved in effects on cerebral function [7].
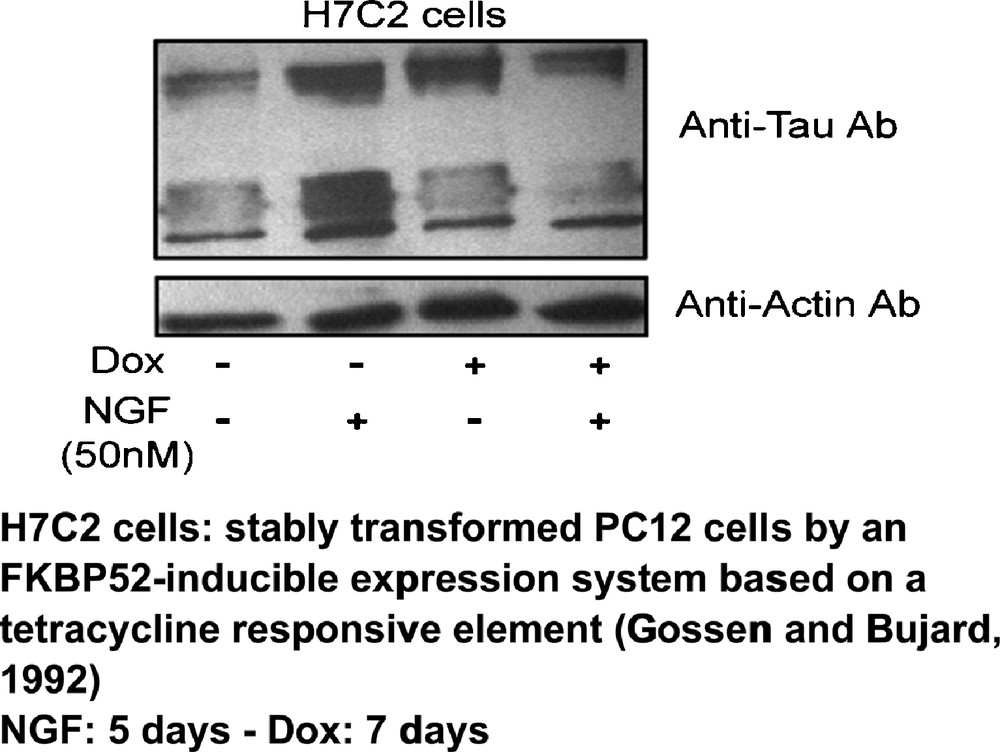
(Color online). Effect of FKBP52 overexpression on tau protein accumulation [5].
We expect to become able to monitor appropriately, in Alzheimer and dementia therapy, the amount/activity of FKBP52. Our current work is involved in the research of ligands of FKBP52 able to eliminate tau protein pathological activity while restoring its physiological function on microtubules (Fig. 3). Of course, considering the variety of pathological tau proteins, it is not yet known whether or not FKBP52 will act as “anti-pathological tau” in all cases. Presently, we include in our current studies mass-spectrometry analyses of FKBP52–tau interaction (with Dr. G. Lippens, CNRS in Lille), lysosomal metabolism of FKBP52 (we just have described the interaction of FKBP52 with lysosomes of Alzheimer brains [14]), and the lentivirus technology to study the appropriate proteins [5]. We also continue to systematically measure FKBP52 in the Central Spinal Fluid (CSF) in order to assess the biological and medical importance of this protein as a new biomarker (collaboration with Drs. N. Lahlou and M. Roger, Institut Cochin, and Drs. C. Hirtz and J. Vialaret, CHU Montpellier).
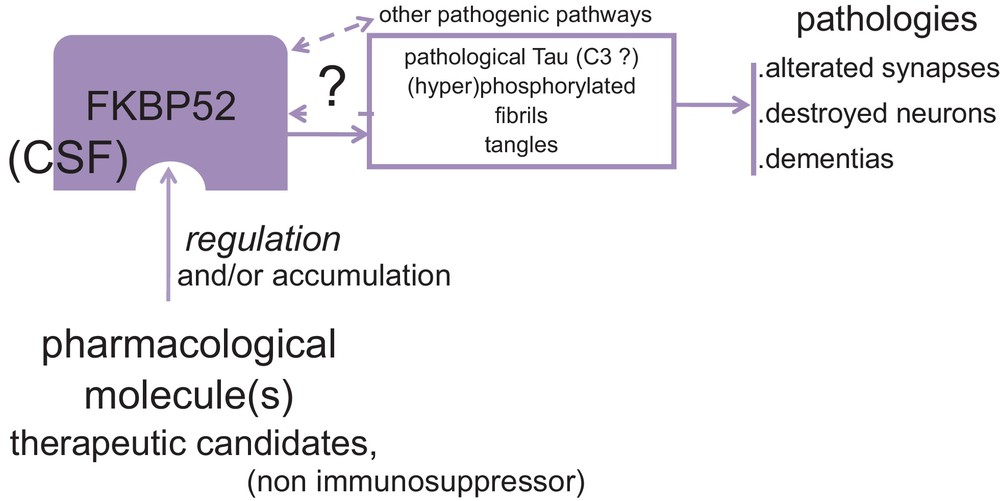
(Color online). Summary of the work hypothesis and strategy for prevention/treatment of dementias via the novel target FKBP52.
Disclosure of interest
The author declares that he has no conflicts of interest concerning this article.