

1 Introduction
The early transverse redistribution of plasma membrane phosphatidylserine (PS) is one of the well-documented hallmarks of cells undergoing apoptosis. The loss of phospholipid asymmetric distribution precedes the loss of plasma membrane integrity 〚1〛. The resulting exposure of PS in the outer leaflet may serve as a recognition signal for phagocytosis of senescent cells to be rapidly cleared 〚2, 3〛. A variety of studies point to the role of Ca2+ in the regulation of apoptosis, there is however no general model explaining the involvement of Ca2+ in apoptosis. In the early stages of apoptosis, changes in cytosolic Ca2+ seem to play a critical role by triggering the activation of Ca2+-dependent events inducing global intracellular and morphological modifications 〚4, 5〛.
Tissue factor (TF), in association with activated factor VII (FVIIa) at the procoagulant membrane surface, initiates the coagulation cascade 〚6–9〛. However, the sole presence of TF protein is not sufficient to bring the process to completion. PS has been shown to enhance the activity of TF 〚6, 9–11〛. In a previous study, we have developed a model of K562 cells transfected with human TF cDNA and with a preserved reduced capacity to expose PS. We have also reported the specific and essential contribution of externalised PS in the development of TF clotting activity at the cell surface 〚12〛. Although K562 cells are refractory to expose PS after procoagulant stimulation, a recent study has shown that apoptosis induced by differentiation leads to PS externalisation 〚13〛.
FVIIa is the only known ligand for TF and its interaction with TF at the cell surface triggers intracellular signalling events. The mechanisms for FVIIa-induced signal transduction are however not completely known. Several studies speculate for a possible mechanism of action of FVIIa independent of TF 〚14〛. In a human keratinocyte cell line, constitutively expressing TF, a group has reported signal transduction induced via the binding of both FVIIa (100 nM) and FXa (174 nM) and has suggested the involvement of protease-activated receptors (PARs) in this particular transduction pathway 〚14〛. However, the levels of the corresponding circulating (inactivated) clotting factors in plasma are indeed lower than those used in this study, with respect to the fact that concentration of FVII in healthy subject is ∼10 nM and that of FVIIa is only ∼0.1 nM 〚15〛. Furthermore, the concentration of generated FVIIa during an apoptotic process is unknown. Other authors have found that recombinant FVIIa (rFVIIa) may activate sufficient amount of FX on activated platelets to promote significant thrombin generation 〚16〛. They speculate that localisation of rFVIIa activity at the activated platelet surface can explain both its safety and efficacy, as well as its haemostatic effect in patients with thrombocytopenia or platelet dysfunctions. In vivo, activated platelets are the main source of procoagulant anionic phospholipids, in particular PS, exposed at their surface and that of derived membrane microparticles, and thus provide the appropriate catalytic surface for the calcium-dependent binding of circulating vitamin K-dependent factors through their γ-carboxyglutamyl-rich N-terminal extremity 〚17–19〛. Being one of the latter, FVIIa may therefore act via the binding of its Gla domain on anionic aminophospholipids exposed at the cell surface after procoagulant stimulation or induction of apoptosis.
This prompted us to assess possible effects of FVIIa on the membrane features of induced cell death in K562 and derived transfected cells, the latter expressing TF.
Abbreviations used in this study are: 〚Ca2+〛i, cytosolic free calcium concentration; FII, prothrombin; FVIIa, activated factor VII; FX, factor X; Fluo-3/AM, fluo-3/acetoxymethyl ester; Gla domain, γ-carboxyglutamic domain; PS, phosphatidylserine; rFVIIa, recombinant activated FVII; TF, tissue factor; z-VAD.fmk, z-Val-Ala-Asp.fluoromethylketone.
2 Materials and methods
2.1 Reagents
RPMI 1640, foetal calf serum (FCS) and geneticin (G418) were from Life Technologies (Paisley, UK); other cell culture reagents were from BioWhittaker (Walkersville, MD). Fluo-3/acetoxymethyl ester (Fluo-3) was from Alexis Corp. (San Diego, CA). z-VAD.fmk (z-Val-Ala-Asp.fluoromethylketone) was from Calbiochem (La Jolla, CA). Recombinant activated human factor VII (rFVIIa) was a kind gift from Novo Nordisk Pharmaceutique SA (Boulogne-Billancourt, France). Propidium iodide (PI), type I-A RNAse A, hirudin and hemin were from Sigma Chemical Co. (St. Louis, MO). Human blood coagulation factor prothombin was the same as that used previously in our laboratory 〚12〛.
2.2 Cell culture and induction of apoptosis
K562 is a highly undifferentiated lineage (ATCC CCL-243) isolated from a Caucasian human with chronic myelogenous leukemia. K562 cells were seeded at 1 × 105 cells ml–1 and cultured in RPMI 1640 supplemented with L-glutamine (2 mM), 1% (v/v) non-essential amino acids, sodium pyruvate (1 mM), gentamicin (20 μg ml–1) and 10% (v/v) heat-inactivated FCS, at 37 °C in humidified 5% CO2 atmosphere. In a previous study, K562 cells, which do not express TF and show a reduced ability to externalise PS, have been used for transfection purpose with human TF cDNA. We obtained one clone expressing TF (DC9) and another (AG8) clone, transfected with the vector alone, was used as control 〚12〛. These clones were cultured in complemented RPMI 1640 containing 0.8 mg ml–1 of geneticin (G418). The cell viability was checked by Trypan blue exclusion. Cells were seeded at 5 × 105 cells ml–1 in the presence or in the absence of hemin (50 μM) for 12 or 24 h.
2.3 Determination of hypodiploid DNA
After the different treatments, cells were harvested and numbered. Concentration in a 70% ethanol solution in H2O was adjusted to 5 × 105 cells ml–1 and fixation was allowed to proceed during at least 1 h at 4 °C. Cells were washed once in Hank’s Balanced Salt Solution (HBSS) before resuspension in a solution containing type I-A RNAse A (0.5 mg ml–1) in HBSS and incubation for 10 min at 37 °C. PI was added at a final concentration of 0.1 mg ml–1. Samples were allowed to stand for another 15 min in the dark at room temperature before flow cytometry analysis using CELLQuest software (Becton Dickinson, San Jose, CA).
2.4 Measurement of 〚Ca2+〛i by flow cytometry
K562 cells were studied at a concentration of 106 cells ml–1. Cells were loaded with 3 μM Fluo-3/AM for 30 min at room temperature. Cells were then washed once in RPMI 1640. Then 10-μg ml–1 PI was added in order to determine the proportion of necrotic cells showing PI uptake. Fluo-3 and PI fluorescence were recorded using a FACScan Becton-Dickinson flow cytometer and the CELLQuest software to analyse about 10 000 events per determination. Fluo-3 fluorescence was plotted as FL-1 versus PI fluorescence as FL-2, and both were measured in fluorescence arbitrary intensity units.
2.5 Statistical analysis
Results are expressed as mean ± SEM of at least three separate experiments performed at different culture stages and unpaired student’s t-test was used for the statistical analysis. A p < 0.05 value was considered significant.
3 Results
3.1 Effect of activated FVII on apoptosis
As shown in Fig. 1, hemin (50 μM) induced apoptosis to the same extent in K562 cells and in transfected DC9 cells, a derived clone expressing TF after transfection 〚12〛, as assessed by determination of hypodiploid DNA (16 ± 1.4% and 15.6 ± 1.3% respectively). AG8 cells, a clone containing the pcDNA3 vector alone, were used as control. At the same concentration, hemin treatment resulted in a weaker increase in apoptosis in these cells (9 ± 1%) (Fig. 1B).
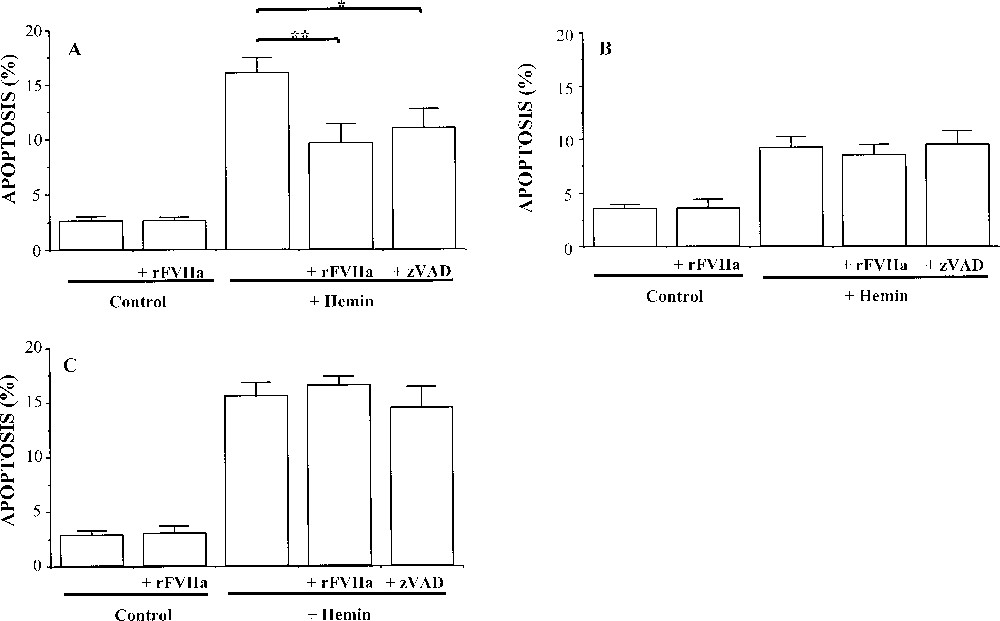
Histograms showing the effect of recombinant activated human FVII (rFVIIa) on hemin-induced apoptotis of (A) K562 cells, (B) transfected K562 cells, AG8 control clone (vector alone), and (C) K562 cells transfected for tissue factor (TF) expression, DC9 clone. The cells were treated with either hemin (50 μM), hemin + rFVIIa (50 nM) or hemin + zVAD (50 μM) for 24 h. Values are mean ± SEM of at least eight independent determinations. ** p < 0.01 indicates a degree of apoptosis significantly different from that observed in the absence of rFVIIa, and * p < 0.05 in the absence of ZVAD.
To examine the possible effect of rFVIIa on the apoptotic process, hemin-treated cells were maintained in the presence or absence of 50 nM rFVIIa during the whole course of the 24-h apoptogenic treatment. rFVIIa treatment significantly reduced hemin-induced apoptosis only in K562 cells (∼40% of inhibition, p < 0.01). It should be noted that rFVIIa alone (in the absence of hemin) did not have any influence on all cell types. These results suggest that the effect of rFVIIa on apoptosis is probably independent of the binding to its TF receptor.
The caspase inhibitor z-VAD.fmk (50 μM) prevented hemin-induced apoptosis in K562 cells (∼25% of inhibition, p < 0.05), suggesting that differentiation-dependent apoptosis is a caspase-dependent event. However, no inhibition by z-VAD.fmk was detected in apoptotic DC9 and AG8 clone.
Since AG8 cells were more resistant to hemin treatment and FVIIa had no significant effect on apoptosis on DC9 and AG8 clones, only K562 cells were selected for the next investigations.
3.2 Involvement of Ca2+ in the effect of rFVIIa
To study the mechanisms potentially responsible for the inhibition by rFVIIa of hemin-induced apoptosis of K562 cells, Ca2+ signalling was investigated in viable cells using fluorochrome Fluo-3 and PI. All experiments were performed in the presence of Fluo-3 and PI, but only the PI negative cell population was gated. Fig. 2 shows the analysis of Fluo-3/PI fluorescence variations by flow cytometry in control cells.
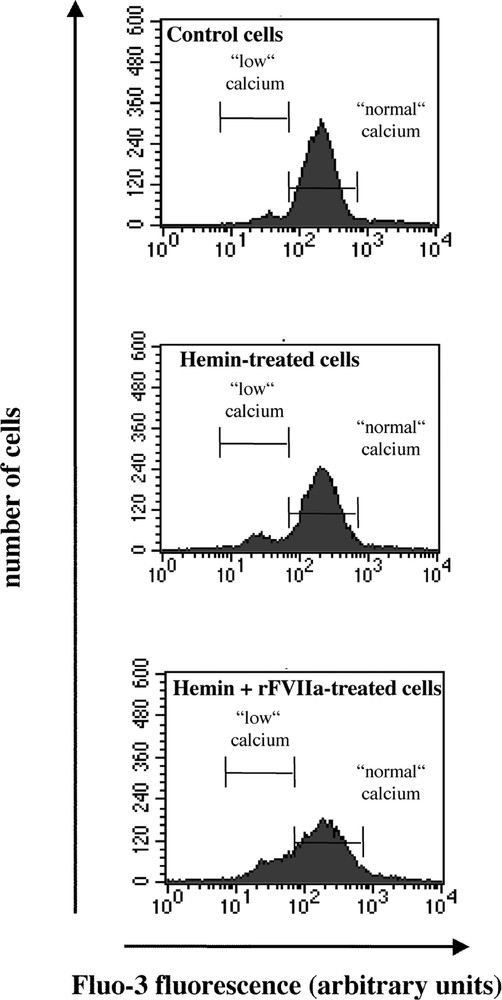
Flow cytometric analysis of hemin and rFVIIa-treated K562 cells. Cells were loaded with the fluorochrome Fluo-3 (3 μM) and propidium iodide (PI, 10 μg ml–1), and gated for PI negative population. Histograms showing the number of cells as a function of Fluo-3 fluorescence. Cells were treated with hemin (50 μM) or FVIIa (50 nM) + hemin for 24 h. One representative experiment of five performed likewise is shown.
Under control conditions (n = 5) (Fig. 2), approximately 92% of cells presented ‘normal’ 〚Ca2+〛i (215 ± 20 fluorescence arbitrary units) and only 2.6 ± 0.8% of cells showed a ‘low’ 〚Ca2+〛i. After induction of apoptosis by hemin (Table 1), the latter population increased in proportion. rFVIIa treatment was responsible for an increase in both the number of cells with ‘low’ Ca2+ and fluorescence intensity itself (p < 0.05).
Effects of rFVIIa on Fluo-3 fluorescence in K562 cells. Results are expressed as the mean ± SEM of Fluo-3 fluorescence intensity. §: Proportion of cells ± SEM with low 〚Ca2+〛i, as shown in Fig. 2. Data are from five independent experiments. * p < 0.05: versus hemin-treated cells; ** p < 0.01: versus control.
| Treatment | Fluo-3 fluorescence intensity | % cells § with low 〚Ca 2+ 〛 i |
| (arbitrary units) | ||
| Control | 53.55 ± 2.7 | 2.6 ± 0.8 |
| Hemin | 29.2 ± 2.5 ** | 8.16 ± 1.6 |
| Hemin + rFVIIa | 41.1 ± 5.1 * | 12.8 ± 3.8 |
| Hemin + zVAD | 39.9 ± 6.1 | 7.4 ± 1.6 |
As shown in Fig. 1, z-VAD.fmk (50 μM) prevented hemin-induced apoptosis in K562 cells. Accordingly, no change in both 〚Ca2+〛i and number of events was observed for hemin + z-VAD.fmk-treated cells (Table 1).
These results suggest that rFVIIa prevents apoptosis by a mechanism probably linked to the maintenance of Ca2+ homeostasis in K562 cells.
3.3 Effect of prothrombin on apoptosis and 〚Ca2+〛i
To establish the significance of the effect of rFVIIa on apoptosis, we examined the role of another vitamin K-dependent clotting factor, prothrombin, possessing a γ-carboxyglutamic (Gla) domain highly homologous to that of FVIIa (data not shown). Prothrombin also reduced hemin-induced apoptosis of K562 cells (58% of inhibition), to the same extent as rFVIIa. Hirudin did not modify the response of apoptotic K562 cells in the presence of prothrombin, testifying to the absence of effect of thrombin eventually generated at the cell surface. This suggests that both vitamin K-dependent factors exert an anti-apoptotic effect by a mechanism involving their Gla-domain.
4 Discussion
It is shown here that recombinant human FVIIa is able to modulate hemin-induced apoptosis of K562 cells, which do not express TF, the receptor of FVIIa. This TF-independent effect was also observed with prothrombin, another vitamin K-dependent coagulation factor that likewise FVIIa possesses a Gla domain. In addition, rFVIIa treatment diminished the alterations of Ca2+ signalling observed following hemin-induced apoptosis, which may explain, at least in part, the anti-apoptotic effect of rFVIIa.
We have previously established a cellular model allowing the assessment of the role of the lipid environment on TF activity with respect to PS accessibility 〚12〛. We have observed that the availability of PS at the membrane surface is critical for TF activity. However, another group has shown that K562 cells can expose PS during apoptosis-induced differentiation 〚13〛, suggesting that PS exposure in the procoagulant response or during apoptosis is regulated by different pathways 〚20〛. Here, we took advantage of hemin ability to induce apoptosis during differentiation of K562 cells and derived transfected clones. For unknown reasons, AG8, the clone containing the vector alone, was more resistant to hemin treatment than both the native K562 and TF-transfected DC9 cells. Under these conditions, the TF-independent effect of FVIIa can be masked by the resistance to apoptosis.
Surprisingly, rFVIIa had no effect on apoptosis of DC9 cells, despite the fact that these transfectants express TF at their surface. Under the same conditions, this difference suggests that FVIIa probably acts via a mechanism independent of TF. The hypothesis is in agreement with other studies, providing evidence for an effect of FVIIa independent of TF. High concentrations of recombinant activated FVII have been found to be efficient in promoting haemostasis in haemophiliacs and in individuals with acquired inhibitors to FVIII or FIX 〚21, 22〛, but the underlying mechanism of this therapeutic property remains unclear. Does high-dose rFVIIa therapy primarily work by a TF-dependent or -independent mechanism? Monroe et al. 〚22〛 have observed that, at concentrations comparable with those attained therapeutically, rFVIIa activates enough FX on stimulated platelets to induce platelet surface thrombin generation. These data lead to consider a TF-independent mechanism for the haemostatic effect of high-dose rFVIIa 〚16〛. Furthermore, intracellular signalling induced by coagulation factors VIIa and Xa in human keratinocytes is linked to an elevation of cytosolic Ca2+, activation of MAP kinase pathway through phosphorylation of extracellular signal-regulated kinase (ERK), p38 and c-Jun N-terminal kinase, and up-regulation of transcription of early growth response gene-1 (egr-1) 〚14〛. These authors have proposed a dual combination for these effects: (i) FVIIa could directly activate its receptor (TF-dependent component), or (ii) protease-activated receptors (PARs) activation may be involved (TF-independent component) in response to FVIIa and FXa.
Another possible explanation is that FVIIa interacts with the procoagulant phospholipids exposed at the surface of the apoptotic membrane through its Gla domain. In this respect, prothrombin is also able to reduce hemin-induced apoptosis in K562 cells, to the same extent as FVIIa. Likewise FVIIa, prothrombin, an ‘inactive’ pro-enzyme, could act through the formation of a sort of ‘exoskeleton’ due to its ability to bind anionic phospholipids exposed after apoptosis. In a previous study from our group, similar results were obtained with annexin V, a protein with high affinity for PS 〚23〛. Annexin V delayed apoptosis of CEM T cells by exerting an external constraint at the plasma membrane. It is well established that the intracellular events leading to death are linked to the external membrane remodelling occurring during apoptosis 〚24, 25〛. Further experiments using other vitamin K-dependent clotting factors lacking the Gla domain (Gla-domainless factors) or rFVIIa inhibited at the active site should certainly be of interest for a better understanding of the mechanism of action of FVIIa on apoptosis.
Induction of apoptosis by hemin produced a moderate, but significant, decrease in 〚Ca2+〛i. rFVIIa significantly restored 〚Ca2+〛i close to the normal level of control cells and counteracted apoptosis. Although, in the presence of rFVIIa, the number of cells with ‘low’ Ca2+ represents a small cell population, the resulting sustained elevation of 〚Ca2+〛i may account for the reduction of the degree of apoptosis. However, in the presence of hemin and rFVIIa, the number of cells with ‘low’ Ca2+ is not significantly different from that measured in the presence of hemin alone. It is generally agreed that variations in 〚Ca2+〛i due to modifications of intracellular Ca2+ stores and/or to Ca2+ influx from the extracellular medium are associated with induction of apoptosis, independently of the apoptogenic stimulus. However, the effect of Ca2+ on apoptosis strongly depends on the cell type. Some cell types seem to be protected from apoptosis by Ca2+ influx 〚26–30〛. In this regard, the maintenance of 〚Ca2+〛i induced by rFVIIa could exert a protective effect towards differentiation-induced apoptosis of K562 cells. Complementary experiments are however necessary to further characterise the underlying mechanism.
5 Conclusion
In conclusion, these findings indicate that rFVIIa evokes a protective response in apoptotic K562 cells, which requires its Gla domain and implicates Ca2+ signalling. Now, the issue raised by this study is that of the possible impact of treatment by high-dose rFVIIa on the prolonged survival of certain cell types to be normally eliminated by apoptosis, including autoreactive B lymphocytes or potentially tumoregenic cells, for instance. Here, an in vitro rFVIIa concentration of 50 nM was used throughout the study. Although the equivalent may be reached in vivo just after rFVIIa injection, it is only transient. Moreover, the prothrombin physiological concentration is in the 1-μM range, making it a candidate more susceptible to interfere in apoptosis of cells within the vascular compartment through the conserved Gla domain of the vitamin K-dependent clotting factors. This does not exclude, however, a specific role of FVIIa with respect to its catalytic activity, which must be further investigated.
Version abrégée
L’exposition de phosphatidylsérine (PhtdSer) est une caractéristique précoce et générale des cellules stimulées ou entrant en apoptose ; lorsqu’elle est combinée à l’expression du facteur tissulaire (FT), elle peut être responsable d’un potentiel thrombogène accru. Par ailleurs, de nombreuses études mettent en évidence un rôle du FT en tant que récepteur capable d’induire une signalisation intracellulaire après liaison de son ligand, le facteur VII activé. C’est dans ce contexte que s’inscrit notre hypothèse de travail, qui suppose que le FVIIa serait capable de promouvoir un effet cellulaire, en interagissant, peut-être via son domaine dit Gla (riche en résidus d’acide γ–carboxyglutamique), avec les phospholipides anioniques, en particulier la PhtdSer, exposés en réponse à une stimulation procoagulante ou, dans notre cas, à un processus d’apoptose. Dans une étude précédente, nous avons développé un modèle de cellules érythroleucémiques K562 transfectées avec l’ADNc du FT humain, tout en préservant une capacité réduite à exposer la PhtdSer. Nous avons alors montré le rôle spécifique et essentiel de la PhtdSer externalisée dans le développement de l’activité procoagulante du FT à la surface cellulaire. Alors que les cellules K562 sont relativement réfractaires à l’expression de PhtdSer après stimulation procoagulante, une étude récente a montré que l’apoptose induite par différenciation à l’aide de l’hémine conduit à l’exposition de PhtdSer.
Nous montrons ici que le FVIIa recombinant est capable de moduler l’apoptose induite par l’hémine des cellules K562 qui n’expriment pas le FT, le récepteur du FVIIa. Ces résultats suggèrent un rôle du FVIIa indépendant de son interaction avec le FT. Par ailleurs, des résultats préliminaires similaires, obtenus avec la prothrombine, un autre facteur de la coagulation vitamine K-dépendant, viennent confirmer ces dernières données. L’induction de l’apoptose des cellules K562 provoque une diminution du Ca2+ cytosolique et l’effet anti-apoptotique du FVIIa passerait par une augmentation du niveau de 〚Ca2+〛i, la ramenant à un niveau proche de celui des cellules non traitées. Le mécanisme par lequel le FVIIa exerce cet effet cellulaire reste à définir, mais on peut d’ores et déjà penser qu’il implique le domaine Gla.
La plupart des études considèrent le FT comme un récepteur capable d’induire une signalisation intracellulaire après liaison de son ligand, le FVIIa. Cependant, il semble que le FVIIa soit capable d’initier une signalisation cytosolique indépendamment du FT. En effet, le traitement à fortes concentrations de FVIIa suffit à promouvoir une hémostase chez des sujets hémophiles et des patients présentant des inhibiteurs des facteurs VIII et IX, mais les mécanismes conduisant à cet effet thérapeutique restent inconnus. D’après d’autres études, le FVIIa activerait suffisamment de FX à la surface des plaquettes stimulées pour induire la génération de thrombine, suggérant donc un mécanisme indépendant du FT. Par ailleurs, dans des kératinocytes, les facteurs VIIa et Xa sont responsables de l’augmentation de la 〚Ca2+〛i, de l’activation de la voie de plusieurs protéines kinases et de la surexpression de certains gènes. Ces auteurs proposent une combinaison de ces effets : (i) le FVIIa pourrait agir directement sur son récepteur, le FT, et/ou (ii) activer les protease-activated receptors (PARs).
Une autre explication réside dans l’interaction du FVIIa avec les phospholipides procoagulants exposés à la surface des cellules stimulées ou apoptotiques via son domaine Gla. Cette hypothèse semble plausible au vu des résultats obtenus avec la prothombine, qui restent néanmoins à confirmer. En effet, la prothrombine est également capable de diminuer le taux d’apoptose des cellules K562, induite par traitement à l’hémine. Ces résultats suggèrent qu’une contrainte externe exercée sur la membrane plasmique serait capable de moduler le déroulement du processus apoptotique. L’interaction du domaine Gla de ces facteurs avec les phopholipides anioniques exposés à la surface de la cellule apoptotique « figerait » la membrane comme s’il y avait formation d’un exosquelette. Dans notre laboratoire, nous avons suggéré que la formation d’un exosquelette par l’annexine V, en se liant aux phospholipides procoagulants, en particulier la PhtdSer et la PhtdEth, retarde significativement l’apoptose de cellules lymphocytaires T humaines en culture.
Nous avons montré que le FVIIa restaure la 〚Ca2+〛i proche du niveau basal des cellules non traitées et, de ce fait, contrecarre l’apoptose. Les effets du FVIIa observés sur la 〚Ca2+〛i, bien que significatifs, ne concernent cependant qu’une proportion de la population totale. Actuellement, les variations de la 〚Ca2+〛i sont associées à l’induction d’apoptose, indépendamment du stimulus apoptogène. Cependant, l’effet de telles variations calciques sur l’apoptose est fortement lié au type cellulaire, où il peut être tantôt protecteur, tantôt inducteur. Dans ce contexte, le maintien de l’homéostasie calcique par le FVIIa protègerait les cellules K562 de l’apoptose induite par différenciation.
En conclusion, ces résultats indiquent que le FVIIa exerce un effet protecteur sur les cellules K562 apoptotiques, impliquant son domaine Gla et la signalisation calcique. Ceci soulève la question d’un impact potentiel du traitement à fortes doses de FVIIa sur la survie de certains types cellulaires normalement éliminés par apoptose, en particulier les lymphocytes B autoréactifs ou les cellules potentiellement tumorigènes. Dans cette étude, nous avons utilisé une concentration in vitro de FVIIa de 50 nM, ce qui peut correspondre, de façon transitoire, à une injection in vivo de FVIIa. De plus, la concentration physiologique de la prothrombine de l’ordre du μM rend celle-ci susceptible d’interférer dans l’apoptose des cellules du compartiment vasculaire via son domaine Gla, caractéristique des facteurs vitamine K-dépendants. Ceci n’exclut pas pour autant un rôle spécifique du FVIIa au regard de son activité catalytique, mais ce point reste à élucider par des études complémentaires.