

1 Introduction
Three thousand louse species have been described [1] and all are classified within the order Phthiraptea, which is divided into four groups: Rhyncophthirina, Amblycera, Ischocera and Anoplura (or sucking lice) [2,3]. Anoplura are obligate permanent hematophagous parasites of mammals and over 500 species are recognized parasites of all major groups of eutherian mammals, including humans [4]. It is believed that mammalian hosts and their parasite lice have coevolved since the Late Cretaceous period. Indeed, lice have frequently been used as a paradigm of host–parasite coevolution, as louse species are associated with specific hosts [5,6]. Currently, three louse species are specifically associated with humans and have been described according to their location on humans. The pubic or crab louse, Phthirus pubis, belongs to the Phthiridae family, is found on the pubic hair and is physically different from the other human lice [7]. The Pediculidae family comprises the body louse and the head louse. The head louse, Pediculus humanus capitis, lives on the head hair whereas the body louse, Pediculus humanus humanus lives in unwashed clothes. Lice have been recognized as human parasites for some thousands of years [8–19]. Whether modern human pediculidae evolved from a single ancestor associated with early hominidae and spread with major expansions of human populations [20] is likely but not demonstrated. It has been suggested that the head louse is the ancestor form that subsequently invaded clothing [21]. Anatomic differences characterize head or body lice [21] and some authors have also suggested that different strains of head lice evolved in different parts of the world following geographical isolation [3] that are closely associated with hosts of particular racial origin [22,23]. However, other authors have proposed that these differences were due to distortion of exoskeleton during the mounting of specimens [24]. A controversy exists on the monophyletic origin of body or head lice. Do body and head lice belong to a single species or not?
To date, few genes or intergenic sequences have been studied for sucking lice: cytochrome oxidase I (COI) [25], cytochrome b (Cytb) [26], elongation factor-1α (EF-1α) [27], 12S rRNA [28], 18S rRNA [29], and two internal transcribed sequences (ITS1 and 2) [30]. Among these genes, the 18S rRNA, EF-1α and COI have demonstrated to be valuable phylogenetic tools for Phthiraptera [27,29]. Using COI sequences, Leo et al. have suggested that human head and body lice are conspecific rather than issued from monophyletic lineages [25]. However, this analysis was based on the study of only one gene, which thus may not necessarily represent the true phylogeny of these lice. In order to estimate the phylogenetic organization of human pediculidae and determine whether head and body lice belong to the same species, we partially sequenced the 18S rRNA of 155 Pediculus lice from different geographical areas and then the EF-1α and COI genes from 20 of these lice.
2 Material and methods
2.1 Louse collection
One hundred and fifty five adult human lice from 13 geographical origins were included in the 18S rRNA study. In order to validate the phylogenetic organization obtained from the 18S rRNA analysis, we sequenced the EF-1α and COI genes of 28 lice for which DNA remained. Lice were provided by local physicians who collected them from indigenous patients infested with only one type of lice: either head or body lice. The origin and number of studied lice is detailed in Table 1. Lice were conserved at −20 °C until processed further.
Location and hosts of the lice included in this study
| Country | Type | Host | 18S rRNA GenBank | EF-1 GenBank | COI GenBank |
| accession number | accession number | accession number | |||
| Burundi | Head | 6 from 2 separate hosts | AY236413 | AY239280 | Identical to AY239286 |
| Body | 10 from 3 separate hosts | AF139486 | AY239283 | Identical to AY239286 | |
| Rwanda | Head | 2 from 1 host | AY236415 | AY239281 | Identical to AY239286 |
| Body | 2 from 1 host | AY236412 | AY239284 | Identical to AY239286 | |
| Zimbabwe | Body | 2 from 1 host | AF139482 | AY239282 | AY239285 |
| Algeria | Body | 16 from 6 hosts | AY236411 | AY239275 | Identical to AY239286 |
| Tunisia | Body | 1 from 1 host | AF139488 | ND | ND |
| Peru | Body | 27 from 9 hosts | AF139481 | AY239279 | Identical to AY239286 |
| The Netherlands | Body | 14 from 2 hosts | AY236416 | ND | ND |
| United States of America | Body | 2 from a laboratory colony | AF139480 | AY239277 | Identical to AY239286 |
| Portugal | Head | 10 from 5 hosts | AY236414 | AY239272 | AY239288 |
| France | Head | 2 from 1 host | AY236410 | AY239274 | Identical to AY239286 |
| Body | 9 from 2 hosts | AF139478 | AY239276 | AY239287 | |
| Russia | Head | 1 from 1 host | AF139484 | ND | ND |
| Body | 10 from 2 hosts | AF139479 | AY239278 | AY239286 | |
| China | Head | 25 from 11 hosts | AY236417 | AY239273 | Identical to AY239286 |
| Thailand | Head | 16 from 5 hosts | AY236418 | AY239271 | Identical to AY239286 |
2.2 DNA extraction, PCR amplification and sequencing
DNA was extracted from mechanically crushed lice using Qiagen columns (QIAamp tissue kit; QIAGEN, Hilden, Germany) according to the manufacturer's instructions. A 1472- to 1493-bp fragment of the 18S rRNA gene was PCR-amplified using the 18SAIGD consensus primer determined after alignment of 18S rRNA gene sequences of Drosophila melanogaster (Genbank accession number M21017) and Aedes aegypti (Genbank accession number M95126), and the 18SBI reverse primer described by DeSalle et al. [31]. A 348-bp fragment of the EF-1α gene was amplified using the primers EFl-For3 and EF1-Cho10 [32]. A 524-bp fragment of the COI gene was amplified using the primers C1-J-1718 [33] and C1-N-2191 [25]. All primers used in this study are shown in Table 2. Each PCR was carried out in a Peltier PTC-200 model thermal cycler (MJ Research Inc., Watertown, Mass.). 3 μl of each DNA preparation were amplified in a 50-μl reaction mixture containing 50 pM of each primer, 200 μM (each) dATP, dCTP, dGTP and dTTP (Invitrogen, Gaithersburg, USA); 1U eLONGase (Invitrogen); 2 μl of eLONGase buffer A (Invitrogen) and 8 μl of eLONGase buffer B (Invitrogen). The following conditions were used for amplification: initial 3 min of denaturation at 94 °C, followed by 40 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at various temperatures, as indicated in Table 2, and extension for 1 min at 68 °C. Amplification was completed by holding the reaction mixture for 3 min at 68 °C to allow complete extension of the PCR products. PCR products were purified using QIAquick Spin PCR purification kits (QIAGEN) as described by the manufacturer. Sequencing reactions were carried out using the d-Rhodamine Terminator cycle-sequencing ready reaction with Amplitaq Polymerase FS (Perkin-Elmer, Coignieres, France), as described by the manufacturer. For all PCR products, the sequences of both DNA strands were determined twice. Sequencing products were resolved using an ABI 3100 automated sequencer (Perkin-Elmer). Sequence analysis was performed with the software package ABI Prism DNA Sequencing Analysis Software version 3.0 (Perkin-Elmer).
Primers used in this study
| Gene | Primer name (reference) | Primer sequence (5′–3′) | Annealing temperature‡ |
| 18S rRNA | 18SAIDG∗† | TCTGGTTGATCCTGCCAGTA | 58 °C |
| 18SBI∗† [31] | GAGTCTCGTTCGTTATCGGA | 58 °C | |
| 18SBIDG† | ATTCCGATTGCAGAGCCTCG | ||
| 18SAI3D† | TATTAAAGTTGCTGCGGATA | ||
| 18SBIMR† | GGTTCGGCCTGCTTTAAGCA | ||
| 18SAI4D† | GGAGGTTCGAAGGCGATCAG | ||
| EF-1 | EF1-For3∗† [32] | GGNGACAAYGTTGGYTTCAACG | 60 °C |
| EF1-Cho10∗† [32] | ACRGCVACKGTYTGHCKCATGTC | 60 °C | |
| COI | C1-j-1718∗† [33] | GGAGGATTTGGAAATTGATTAGTTCC | 60 °C |
| C1-N-2191∗† [25] | CCAGGAAGAATAAGAATATAAACTTC | 60 °C |
∗ Primers used for PCR amplification.
† Primers used for sequencing.
‡ Annealing temperature is indicated for primers used for PCR only.
2.3 Sequence analysis
DNA sequences from each gene were aligned using the multisequence alignment software CLUSTAL W, version 1.81 [34]. Percentages of similarity were determined using the MEGA software package. Unrooted phylogenetic trees were obtained from DNA sequences by using the maximum parsimony method (DNAPARS software in the 3.4 version of PHYLIP software [35], distance methods (DNADIST: distance matrix with Kimura 2 parameter [36]; and NEIGHBOR: Neighbour-Joining [37]) and the maximum likelihood method (DNAMLK software in PHYLIP). Bootstrap replicates were performed to estimate the node reliability of the phylogenetic trees obtained by the three methods [38]. The bootstrap values were obtained from 100 trees [39] generated randomly with SEQBOOT and CONSENSE in the PHYLIP software package. Only values above 75 were considered significant. Trees were drawn using the TreeView version 1.5 [40] software. Only the parsimony trees are presented in this article. Using the same methods, we also analyzed various combinations of the three genes, i.e. 18S rRNA+EF-1α, 18S rRNA+COI, EF-1α+COI, and 18S rRNA+EF-1α+COI.
3 Results
3.1 18S rRNA gene sequences
The size of 18S rRNA gene PCR products ranged from 1472 bp (Burundi and Rwanda) to 1492 bp (China) for head lice and from 1472 bp (Burundi and Rwanda) to 1493 bp (Zimbabwe) for body lice. Nucleotide differences were observed at 90 positions among tested lice. When several lice of one type (i.e. head or body lice) from a given country were tested, they exhibited identical DNA sequences. We did not identify any specific sequence signature allowing the discrimination between body and head lice, but rather observed sequence signatures distinguishing sub-Saharan lice from other lice at nine nucleotide positions (Fig. 1a).

Signature mutations in the 18S rRNA (a) and EF-1α (b) gene sequences of Pediculus humanus allowing the identification of head an body lice from sub-Saharan Africa or other areas. 18S rRNA sites were numbered according to the body louse from France and EF-1α sites were numbered according to the alignment of sequences. Nucleotides specific of a given group are underlined.
Among sub-Saharan lice, head lice from Burundi and Rwanda exhibited identical 18S rRNA gene sequences. Body lice from Burundi and Rwanda also exhibited identical 18S rRNA gene sequences but differed at 42 positions (97.2% similarity) from Zimbabwean body lice. Head and body lice from Burundi and Rwanda exhibited nearly identical sequences, with only one nucleotide difference (Fig. 1a).
Among lice from all areas except sub-Saharan Africa, the 18S rRNA gene similarity among head lice ranged from 99.1 to 99.9% and the similarity among body lice ranged from 99.7 to 100%. Head lice differed from body lice at 21 positions (98.6% similarity) (Fig. 1a).
3.2 EF-1α gene sequences
For all 28 lice tested, the size of EF-1α PCR products was 348 bp, of which 116 (33%) were variable and 103 (30%) parsimony informative. When several lice of one type (i.e. head or body lice) from a given country were tested, they exhibited identical DNA sequences. When comparing head and body lice regardless of their geographical origin, we observed no discriminant sequence signature, but rather observed differences distinguishing sub-Saharan lice from others at 67 positions (81% similarity) (Fig. 1b).
Among sub-Saharan lice, head lice from Burundi and Rwanda exhibited identical EF-1α gene sequences. Body lice from Burundi and Rwanda were also identical but differed at one position (99.7% similarity) from Zimbabwean body lice. Head lice from Burundi and Rwanda differed at five positions (98.5% similarity) from body lice from the same countries (Fig. 1b).
Among lice from all areas except sub-Saharan Africa, the EF-1α gene similarity ranged from 95.5 to 99.1% among head lice and from 89.9 to 100% among body lice. Head and body lice differed at 18 positions (94.8%) (Fig. 1b).
3.3 COI gene sequences
The size of COI PCR products was 524 bp for all lice. All sequences were identical except those from the French and Zimbabwean body lice, which exhibited one nucleotide difference by comparison with the other tested lice and the Portuguese head lice, which differed from other lice by 19 bases.
3.4 Phylogenetic analysis
The dendrograms obtained using 18S rRNA sequences with the three different tree-building methods exhibited similar organizations. Two clades were clearly distinct: (i) lice from America, Europe, Asia and North Africa clustered together but divided into two sub-groups, one made of head lice and the other comprising body lice; (ii) lice from sub-Saharan Africa clustered together and Burundian lice also divided into head and body louse subgroups. At the nodes where head lice diverged from body lice bootstrap values were above 75% (Fig. 2). The phylogenetic organization based on EF-1α sequences showed the same organization for lice from America, Europe, Asia and North Africa, which were sub-divided into two sub-groups, one made of head lice and the other comprising body lice. However, within lice from sub-Saharan Africa, body lice from Zimbabwe and Burundi were grouped together in a cluster different from that made of Burundian head lice. Both clades of sub-Saharan lice and lice from other areas were supported by 100% bootstrap values and the nodes where head lice diverged from body lice exhibited bootstrap values above 90% (Fig. 3b). The dendrograms obtained with the three different tree-building methods exhibited similar organizations. The phylogenetic analysis using amino acid sequences derived from EF-1α nucleotide sequences showed organizations similar to those obtained from nucleotide sequences. Due to the conservation of COI sequences, any combination of genes including the COI gene provided both similar organization and similar bootstrap values than without this gene (data not shown). In contrast, the combination of 18S rRNA and EF-1α sequences provided the same phylogenetic classification using all three analysis methods and 18 of the 20 nodes were supported by bootstrap values equal to or greater than 90% (Fig. 3c).
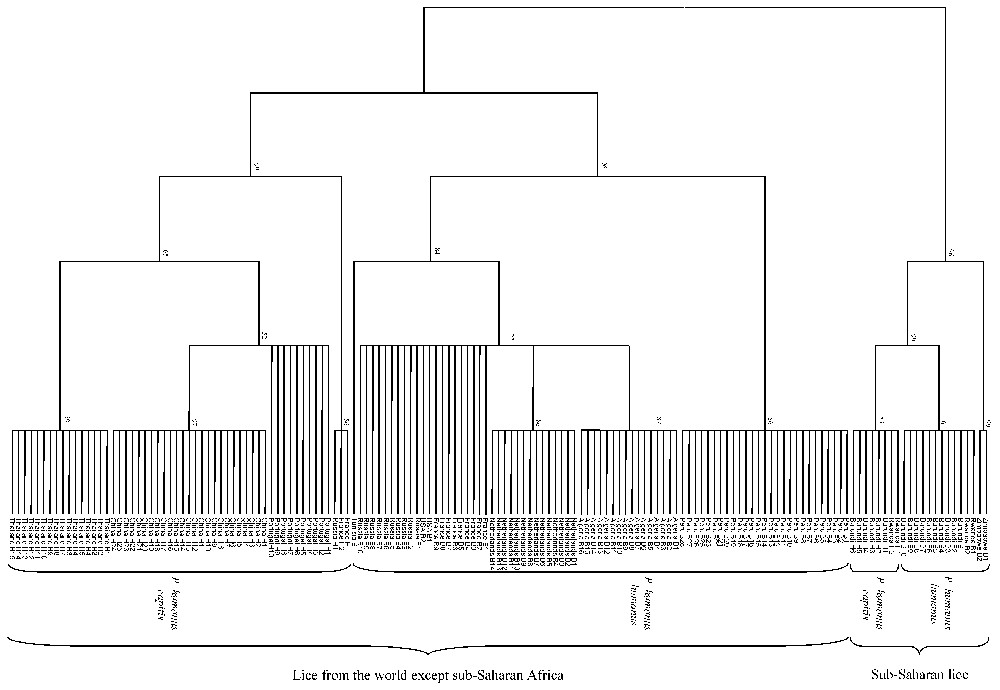
Unrooted cladogram representing the phylogenetic relationships between 155 human lice inferred from the comparison of 18S rDNA sequences using the parsimony method. The numbers at the nodes are the proportion of 100 bootstrap resamplings that support the topology shown. The names of lice appear in the tree as country of origin followed by B for body lice or H for head lice and the louse number.
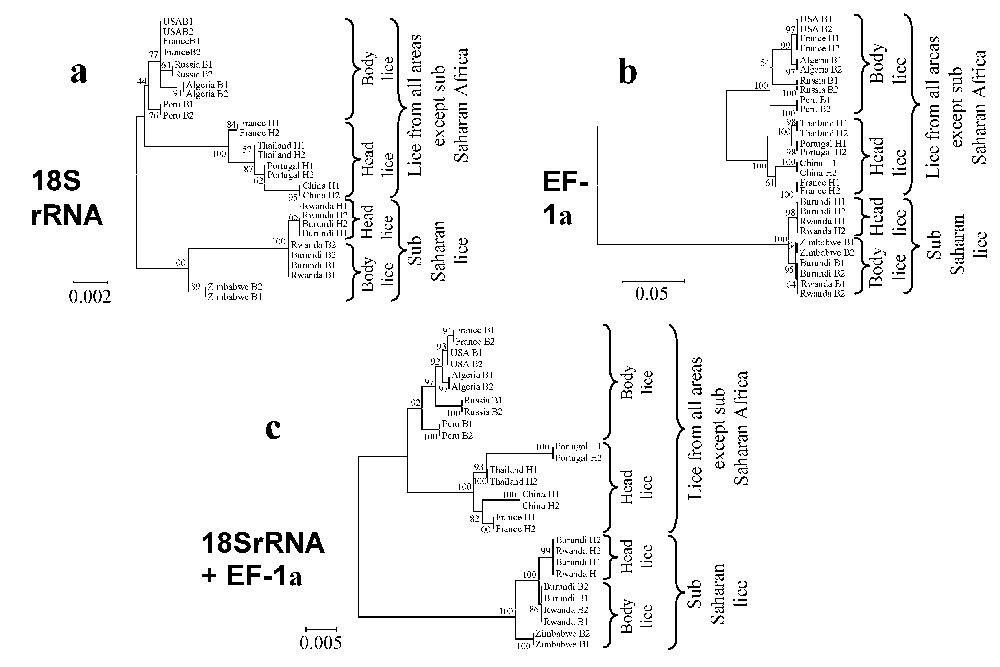
Unrooted cladograms representing the compared phylogenetic relationships between 16 human body and 12 head lice inferred from the comparison of fragments of the 18S rDNA (a), EF-1α (b), and 18S rDNA+EF-1α (c) genes using the Neighbour-Joining method (Kimura 2 parameter). The numbers at the nodes are the proportion of 100 bootstrap resamplings that support the topology shown. The names of lice appear in the trees as country of origin followed by B for body lice or H for head lice and the louse number. The scale bars represent 0.2%, 5% and 0.5% differences in nucleotide sequences for the 18S rDNA (a), EF-1α (b), and 18S rDNA+EF-1α (c) trees, respectively.
4 Discussion
In order to study the phylogenetic organization of Pediculus humanus capitis and Pediculus humanus humanus, we collected 155 human lice from patients suffering only one type of infestation, head or clothing, in order to avoid confusion, as patients who are heavily infested with body lice may also have these ectoparasites on the head. The current taxonomic position of human lice is unclear, as their description and phylogenetic classification have mostly been based on morphological criteria [41–43]. Subtle differences in color and size of different body parts have been reported: head lice were described as being smaller and more heavily pigmented than body lice [3] and as having a shorter femur size [21]. Some authors have reported that the color of head lice may vary according to their hosts and have suggested that it could be the genotypic result of coevolution with different human races [3,22,23], whereas others have proposed that this phenomenon to be only phenotypic, i.e. the darkening of the louse being a form of camouflage which depends on the color of the host's skin and hair [4]. Over recent years, several phylogenetic studies have been conducted on lice, based on the analysis of various genes [25–30]. Among these studies, two have specifically been dedicated to human pediculidae; Leo and Barker, using the ITS2 spacer, have observed that these sequences were present in more than one copy in some lice and were highly variable among lice and even in a single louse, making this tool unsuitable for phylogenetic purposes [30]; the same authors then studied a fragment of the COI gene and suggested, on the basis of the classification of lice from various geographical areas into 10 genotypes due to a high variability of these sequences, that human body and head lice do not represent reciprocally monophyletic lineages and thus, were conspecific [25]. However, these assumptions were based on the study of only one gene and some sequences were obtained from a single DNA strand. As phylogeny based on a single gene may not represent the species phylogeny, due to various biological processes such as hybridization [44,45], differences in lineage sorting [46] and gene duplication [47], we selected three genes, i.e. the 18S rRNA which has previously been used in a multiple gene phylogenetic study of Isochnera [29], the EF-1α, which has proven to be useful to infer the phylogenetic relationships among major groups of lice [27,29], and the COI gene, which had previously been used for human lice [25]. This combination of genes had previously been used to infer phylogenetic relationships among Isochnera [29]. A surprise came from the analysis of the COI gene, which we found highly conserved among human lice from various geographical origins, in contrast with the findings of Leo et al. [25], and which was clearly not suitable to infer phylogenetic relationships among these insects. As sequences from each louse were determined four times (i.e. each DNA strand was sequenced twice), we believe that our data are reliable.
Using 18S rRNA sequences, we found no signature mutations differentiating head and body lice regardless of their geographical origin, but rather signature mutations specific for two groups: sub-Saharan lice and lice from other areas (Fig. 1). In addition, within each of these two groups, additional signature sequences supported the separation of head and body lice. The analysis of EF-1α sequences revealed a large variability of this gene among human lice and confirmed the 18S rRNA analysis. Such signature sequences may help scientists to precisely identify human lice. The phylogenetic study using the 18S rRNA gene showed an organization in accordance with the signature mutations observed in the sequences. This organization was also supported by the analysis of the EF-1α gene, which was highly variable among lice and allowed us to infer a strongly supported phylogenetic classification. We observed that the first divergence among lice was between lice from sub-Saharan Africa and those from all other locations as geographically distant as America, Europe, Northern Africa and Asia. European, American and North African lice clustered together with high bootstrap values. African lice were closely related but differed from the other clade. This divergence and heterogeneity had not been expected because it was believed that lice had been homogeneously exchanged on earth through widespread outbreaks. This exchange may have happened between Europe, Asia, America and Northern Africa, as we observed that lice from these areas were closely related. It would be interesting to study the DNA from American mummies' lice [48] to check their phylogenetic position compared to current American lice.
Our data support the divergence of body and head lice to be the result of two distinct events, whereas it was thought that this was a single split [21]. This observation contradicts the current classification into either two species or subspecies, namely Pediculus humanus corporis and Pediculus humanus capitis. It has been suggested that Asia was potentially the source of human lice [4]. Based on our data, this hypothesis appears unlikely. It is likely that hominidae have evolved from ancestors in Africa [20]. If the association of lice with early hominidae occurred as the result of a single event, we speculate that this may have occurred in Africa, with two louse populations having evolved from these ancestor lice, one African louse population associated with sub-Saharan human African populations and another louse population that may have spread with the major expansions of human populations from Africa [20]. Our data also support the hypothesis that the divergence between current head and body lice occurred as the result of two distinct events, one among sub-Saharan lice and one among lice associated with hominidae, who subsequently spread throughout the world.
Finally, we provide a molecular tool for the identification of human lice. In addition, the phenotypic classification within the Pediculidae family into two groups, namely P. humanus capitis and P. humanus humanus, does not match with our genetic data, as these do not support the continued description of two species or subspecies. We believe that the extension of our work to the testing of lice from other sources and the testing of additional genes may contribute to a better understanding of the relationships between humans and their lice.
Acknowledgements
We thank J.B. Ndihokubwayo, J. Kinigi, P.J. Kelly, J.P. Gonzalez, Z.X. Liang, and F. Bacellar for providing us with lice.