

1 Introduction
The immune response in vertebrates is categorized into innate and acquired immunity. Innate immunity is a first-line of defense system against invading pathogens. The main players of innate immunity are phagocytes such as neutrophils, macrophages, and dendritic cells. These phagocytes engulf pathogens, digest and kill them. They can also discriminate between pathogens and self via Toll-like receptors (TLRs) that recognize conserved motifs found in the microorganism but not in the vertebrate. Engagement of the TLRs triggers anti-microbial responses and cytokine production. More importantly, induction of acquired immunity entirely depends on innate immunity. Thus, TLRs play a crucial role in the induction of acquired immunity as well as innate immunity.
2 Mammalian Toll-like receptors
The Toll receptor is a type I transmembrane receptor, evolutionarily conserved, and its homologs are found in insects, plants and mammals. The extracellular domain of Toll receptors contains leucine-rich repeat (LRR) whereas the cytoplasmic domain shows striking homology with that of IL-Rs, and is referred to as Toll/IL-1R (TIR) domain. The LRR domains are composed of different numbers of repeats of 24–29 amino acids in length, and each repeat contains the motif xxLxLxx as well as other conserved leucines. The Toll gene was initially identified as a gene essential for the dorsal-ventral development in the Drosophila embryo. Subsequently Toll signaling has been shown to play an essential role in the immune response to antifungal infection [1]. Toll-mutant flies show compromised induction of antifungal peptide, and are highly sensitive to fungal infection. This finding triggered search for Toll homologues in mammals through EST and genomic sequence database searches. So far, ten members of TLR family have been identified in mammals [2–4]. The essential role of Drosophila Toll in the antifungal response suggested that mammalian Toll-like receptors also participate in innate immunity. In fact, Toll-like receptors have been found to be essential for microbial recognition (Fig. 1).
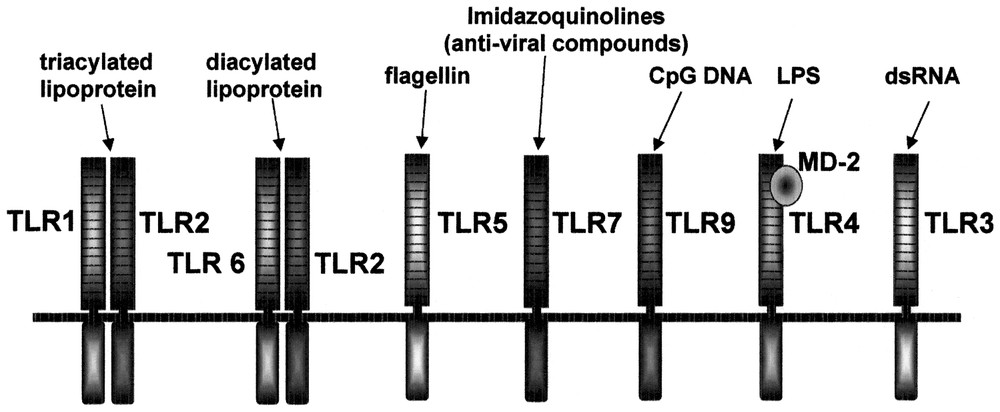
Summary of ligands recognized by TLR family.
2.1 LPS signaling and TLR4
Lipopolysaccharide (LPS) is a component of the outer membranes of Gram-negative bacteria, a potent activator of macrophage and a causative agent of endotoxin shock. Positional cloning of the locus responsible for LPS hyporesponsiveness in C3H/HeJ mice as well as generation of TLR4 knockout mice has demonstrated that TLR4 is essential for LPS signaling [5,6]. However, TLR4 is not sufficient for LPS signaling, but requires an additional molecule, called MD-2 that associates with the extracellular portion of TLR4 [7]. The generally accepted scenario of LPS recognition and signaling in mammals is as follows: LPS released from Gram-negative bacteria is immediately captured by the LPS-binding protein (LBP), a specific lipid transfer protein, and binds CD14 present on the surfaces of mononuclear phagocytes. CD14 lacks a transmembrane domain, and is, thus, incapable of transducing signals. LPS is then delivered to the TLR4/MD-2 complex, which induces dimerization of the TLR4/MD-2 complex and subsequent activation of cytoplasmic signaling molecules.
2.2 Recognition of lipoproteins and peptidoglycan by TLR2
TLR2 recognizes a variety of microbial components. These include peptidoglycan (PGN) from Gram-positive bacteria such as Staphylococcus aureus, lipoproteins/lipopeptides from a variety of bacteria, GPI anchors from Trypanosoma cruzi, lipoarabinomannan from Mycobacterium tuberculosis, porins from Neisseria meningitides, and the yeast cell wall component zymosan [4,8].
2.3 Recognition of lipid configuration of lipoproteins by TLR2/6 and TLR2/1 heterodimers
All membrane-anchored lipoproteins contain a lipolyated amino-terminal residue. The immunostimulatory activity of lipoproteins is attributed to the lipid portion. Fig. 2(a) shows structural difference between MALP-2 and PAM3CSK4. PAM3CSK4 contains a triacylated cysteinyl residue at the N-terminus, whereas the cysteinyl residue in MALP-2 is only diacylated. The response to MALP-2 is completely abolished in both TLR2-deficient and TLR6-deficient macrophages, whereas the response to PAM3CSK4 is normal in TLR6-deficient macrophages in spite of no response in TLR2-deficient macrophages [9]. The replacement of lipid portion of MALP-2 with that of PAM3CSK4 activated TLR6-deficient macrophages, demonstrating that TLR6 discriminates the subtle difference in the lipid portion of these lipopeptides (our unpublished data). Reciprocally, the response to PAM3CSK4 is significantly impaired in TLR1-deficient macrophages while the response to MALP-2 is normal in the mutant cells, indicating that TLR1 preferentially recognizes triacylated configuration of the lipid moiety [10].

Structures of several lipopeptides (a) and recognition of lipoproteins by heterodimers (b).
TLR1-deficient mice responded to PGN, LPS and MALP-2 to the same extent as wild-type mice. As show in Fig. 2(b), MALP-2 (diacylated form) is recognized by heterodimer between TLR2 and TLR6 whereas PAM3CSK4 and N-PAM-S-Lau2CSK (triacylated forms) are more preferentially recognized by heterodimer between TLR2 and TLR1 [10]. The responses to zymosan and Trypanosoma cruzi GPI are also recognized by TLR2/TLR6 heterodimer (our unpublished data). It is possible that TLR other than TLR1 and TLR6 pairs with TLR2 to recognize PGN, or TLR2 alone may suffice to recognize it.
2.4 Double stranded RNA recognition by TLR3
Viral replication within infected cells often results in the generation of dsRNA that can stimulate immune cells. TLR3-deficient mice have shown reduced responses to dsRNA as well as the viral RNA mimic poly (I):poly (C), suggesting that TLR3 is involved in recognition of dsRNA [11].
2.5 Flagellin recognition by TLR5
Flagellin is a 55-kDa monomer obtained from bacterial flagella, a polymeric rod-like appendage extending from the outer membrane of Gram-negative bacteria that propels the organism through its aqueous environment. Flagellin is also a potent proinflammatory factor, the signaling of which has been shown to be mediated through TLR5 [12]. TLR5 is expressed on the basolateral, but not apical, surface of intestinal epithelia [13]. Therefore, flagellin activates proinflammatory gene expression only if it crosses intestinal epithelia and contacts the basolateral membrane, which may in part explain why commensal microbes can secrete flagellin into the intestinal lumen without inducing inflammation.
2.6 Anti-viral chemical compounds and TLR7
TLR7 recognize several types of imidazoquinolines [14]. Imiquimod (Aldara, R-837, S-26308) and R-848 (resiquimod, S-28463) are low molecular weight compounds of the imidazoquinoline family that possess potent antiviral and anti-tumor properties. These imidazoquinolines induce cytokines, including interferon-α and IL-12. Topical imiquimod therapy is now approved for the treatment of external genital and perianal warts caused by Papilloma virus infection. R-848 is a more potent analogue of imiquimod, and trials are underway to assess its use in therapy for herpes virus as well as hepatitis C virus infection. TLR7 recognizes other synthetic chemicals, loxoribine and broprimine as well (our unpublished data).
2.7 CpG DNA and TLR9
TLR9 is essential for the response to bacterial DNA (and viral DNA), as well as synthetic oligodeoxynucleotides containing unmethylated CpG-dinucleotide (CpG DNA) [15]. These oligonucleotides stimulate B cell proliferation and activate macrophages and dendritic cells. The optimal immunostimulatory CpG DNA motifs differ between mouse and human, and this difference has been attributed to the amino acid sequence difference between the extracellular portions of the human and mouse TLR9s [16]. Recent studies indicate that two types of immunostimulatory DNAs exert different immune responses [17,18]. One is CpG DNA with an entirely phosphorothioate backbone, and it stimulates B cell proliferation and induces the production of IL-6 and IL-12 by monocytes. The other is CpG DNA, that contains phosphorothioate G-rich sequences at the ends and a phosphodiester palindromic sequences with a CpG dinucleotide in the middle, and this type of CpG DNA preferentially stimulates type I IFN production by plasmacytoid dendritic cells. In spite of the existence of these two types of CpG DNA with different biological effects, TLR9 KO mice were unresponsive to both, demonstrating that TLR9 is essential to the response to a variety of CpG DNA [19]. One report showed that DNA-PK is also essential for the response to CpG DNA based on the lack of the response to CpG DNA in DNA-PK KO mice [20]. However, DNA-PK-deficient dendritic cells showed upregulation of costimulatory molecules and cytokine production in response to CpG DNA, indicating that DNA-PK is not essential for the response to CpG DNA [21].
3 IL-1R/TLR signaling pathways
IL-1R and TLR family utilize the same signaling molecules. These include the adaptor molecule MyD88, the IL-1R-associated protein kinases (IRAKs), the TGF-β-activated kinase (TAK1), TAK1-binding protein 1 (TAB1) and 2 (TAB2), the tumor necrosis factor receptor-associated factor 6 (TRAF6) [22]. Triggering of IL-1R or TLR recruits the adaptor protein MyD88 to the receptor complex, which in turn enables the association with the IL-1R-associated kinases IRAK-4 and IRAK-1. During the formation of this complex, IRAK-4 is activated, and leads to the hyperphosphorylation of IRAK-1, which then induces interaction with TRAF6. The formation of IRAK-4/IRAK-1/TRAF6 causes conformational changes, and leaves the receptor complex. The IRAK-4/IRAK-1/TRAF6 then interacts with membrane-bound TAB2, and then the TAB2/TAK1/TAB1/TRAF6 complex translocates to the cytoplasm, leaving IRAKs. TAK1 is subsequently activated in the cytoplasm, leading to the activation of IκB kinase (IKK). Phosphorylated IRAK-1 stays at the membrane, and is eventually degraded. Activated IKK causes the activation of NF-κB through the phosphorylation and degradation of IκBs, which sequester NF-κB in the cytoplasms prior to activation. Activation of TAK1 also results in the activation of MAP kinases.
4 MyD88-dependent and -independent pathways in TLR4 signaling
MyD88 is an adapter molecule that functions to recruit IRAK to the IL-1 or TLR4 receptor complexes after IL-1 or LPS stimulation, respectively. Indeed, MyD88-deficient mice are unresponsive to IL-1, LPS and other microbial cell wall components such as peptidoglycan and lipopeptides [23–25]. However, there is a difference in the signaling pathways triggered by LPS and by these latter stimuli. Mycoplasmal lipopeptide activation of NF-κB and MAP kinases, which is mediated by TLR2, is completely abolished in TLR2-deficient or MyD88-deficient macrophages. However, LPS activation of MAP kinases and NF-κB remains intact in MyD88-deficient macrophages, although activation is delayed compared with wild-type mice [24]. This indicates that the LPS response is mediated by both MyD88-dependent and -independent pathways, each of which leads to the activation of MAP kinases and NF-κB. Nevertheless, the MyD88-dependent pathway is essential for the inflammatory response mediated by LPS. In an attempt to characterize the MyD88-independent pathway, we tried to identify the genes induced in a MyD88-independent manner [26]. We generated cDNA library from MyD88-deficient macrophages before and after LPS stimulation and performed a subtractive suppression hybridization. This screening identified several genes induced in LPS-stimulated MyD88-deficient macrophages. Most were a CXC chemokine, IP-10, and others included GARG16 and IRG-1. These are so-called interferon inducible genes. Consistent with the lack of MyD88-independent pathway in the TLR2 signaling, wild-type macrophages did not induce IP-10 mRNA in response to mycoplasmal lipopeptide. It is shown that IFN inducible genes are induced by type I interferons. Analyses with IFN- R KO mice and STAT1 KO mice showed that IP-10 induction is secondary to IFN-β induction in response to LPS [27]. In contrast, GARG16 was induced independently of MyD88 and IFN-β. A recent paper has shown the same conclusion [28].
Previous studies show that interferon inducible genes including IP-10 are regulated by IRF-3. After viral infection, treatment with dsRNA, or LPS stimulation, IRF-3 undergoes phosphorylation and translocates from the cytoplasm to the nucleus, resulting in transcriptional activation of interferon-inducible genes. As expected, the ISRE binding activity and IRF-3 activation were induced in a MyD88-independent manner [24]. Fig. 3 summarizes the difference in the signaling pathway and gene expression between LPS and mycoplasmal lipoprotein.
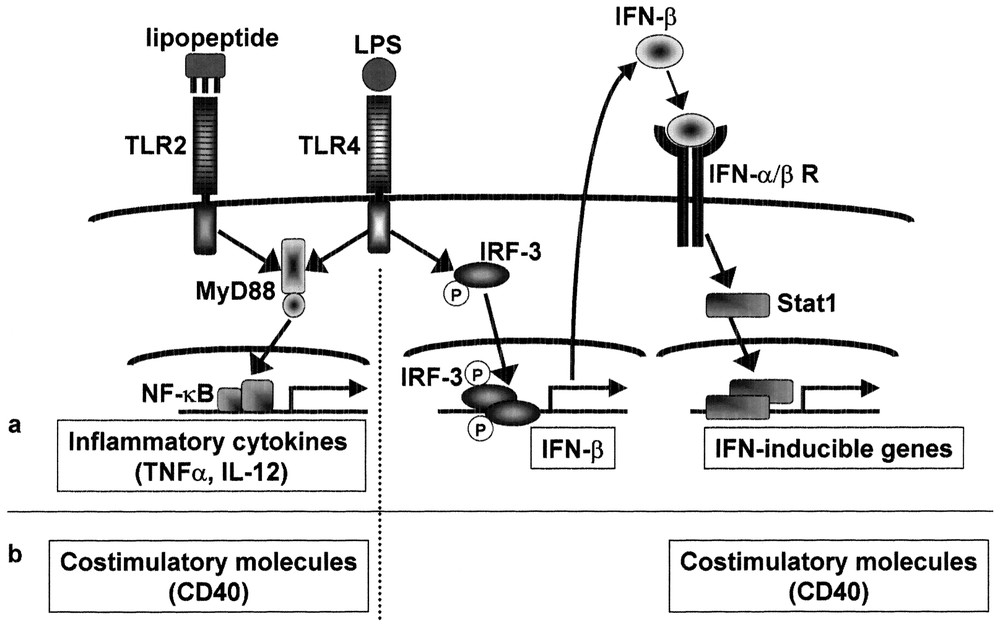
MyD88-independent signaling pathways for induction of IFN-inducible genes and costimulatory molecules. Mycoplasmal lipopeptide induces inflammatory cytokines through TLR2-MyD88-dependent pathway, but does not induce IFN-β or IFN-inducible genes due to lack of MyD88-independent pathway. LPS induces proinflammatory cytokines as well as IFN-β and IFN-inducible genes. The IFN-β gene is probably activated via phosphorylation and nuclear translocation of IRF-3. Induction of IFN-inducible genes is secondary to IFN-β induction and therefore STAT1-dependent. Mycoplasmal lipopeptide induces CD40 through TLR2-MyD88-dependent pathway. LPS induces CD40 either through TLR4-MyD88-dependent pathway or through TLR4-MyD88-independent pathway via IRF-3 activation and subsequent IFN-β induction.
In addition to the induction of IFN-inducible genes, the MyD88-independent pathway is also involved in the LPS-mediated maturation of DCs, which accompanies the induction of costimulatory molecules and production of inflammatory cytokines [29]. When MyD88-deficient bone marrow-derived DCs were cultured with LPS, they augmented surface expression of co-stimulatory molecules such as CD40, CD80 and CD86, and activated T cells to proliferate, whereas TLR4-deficient DCs failed to maturate in response to LPS, indicating that DC maturation proceeds without MyD88 in response to LPS. Further studies showed that activation of either MyD88-dependent or MyD88-independent pathway is enough for induction of costimulatory molecules. Nevertheless, MyD88-independent induction of costimulatory molecules is mainly secondary to IFN-β induction as is the case of interferon-inducible genes [27].
5 Identifiation of other adaptor molecules involved in the TLR signaling
Another TIR domain-containing protein (TIRAP, also named Mal) has been identified by a database search. TIRAP was shown to associate with TLR-4 but not other TLRs [30,31]. Overexpression of a dominant negative form of TIRAP inhibited LPS-induced NF-κB activation or DC maturation. Blockage of TIRAP using a cell-permeable inhibitory peptide also prevented induction of IP-10 by LPS [28]. Thus, it was expected that TIRAP/Mal might participate in the MyD88-independent pathway. However, studies with TIRAP-deficient mice have revealed that TIRAP/Mal is not involved in the MyD88-independent pathway, and acts as an adaptor in the MyD88-dependent signaling pathways via TLR2 and TLR4 [32,33]. Recently, another adaptor molecule named TRIF or TICAM-1 has been identified [34,35]. Overexpression of TRIF, but not MyD88 or TIRAP, preferentially activates the promoter of the interferon β. A dominant negative form (DN) of TRIF, but not MyD88DN or TIRAPDN, blocked poly I:C-mediated TLR3 response, indicating the specific role of this novel adaptor in TLR3 signaling. TRIFDN also blocked the signaling pathways activated by other TLR family, which indicates some role of TRIF in MyD88-dependent pathway. Further analyses including generation of TRIF knockout mice will be required for the precise role of TRIF in the TLR signaling.
TLR3 signaling mainly depends on the MyD88-independent pathway. Poly I:C induce activation of NF-κB and MAP kinases in MyD88-deficient macrophages almost to the same extent as wild-type macrophages (our unpublished data). Dominant negative form of MyD88 or TIRAP/Mal did not abolish the poly I:C-mediated TLR3 response. There is no any interaction detected between MyD88 or Mal and TLR3 upon poly I:C stimulation. Recently, a paper reported that TLR3-mediated activation of NF-κB and MAP kinses is through an IRAK-independent pathway, employing signaling components TLR3-TRAF6-TAK1-TAB2-PKR [36]. TRIF is a likely candidate adaptor to recruit TRAF6-TAK-1-TAB2 to TLR3. In fact, TRIF harbors three TRAF6 binding motifs in the N-terminus. More recently, two papers independently showed that non-canonical IκB kinases, IKKε (also called IKKi) and TBK1 are involved in IRF3 activation, and that they may function downstream of adaptor TRIF, which specifically activates the IFN-β promoter.
A database search of TIR domain-containing adaptors has revealed two additional adaptor molecules (our unpublished data). These findings suggest that several TIR domain-containing adaptor molecules are involved in the TLR-mediated signaling pathways and differential utilization may provides the specificity in the TLR signaling (Fig. 4).
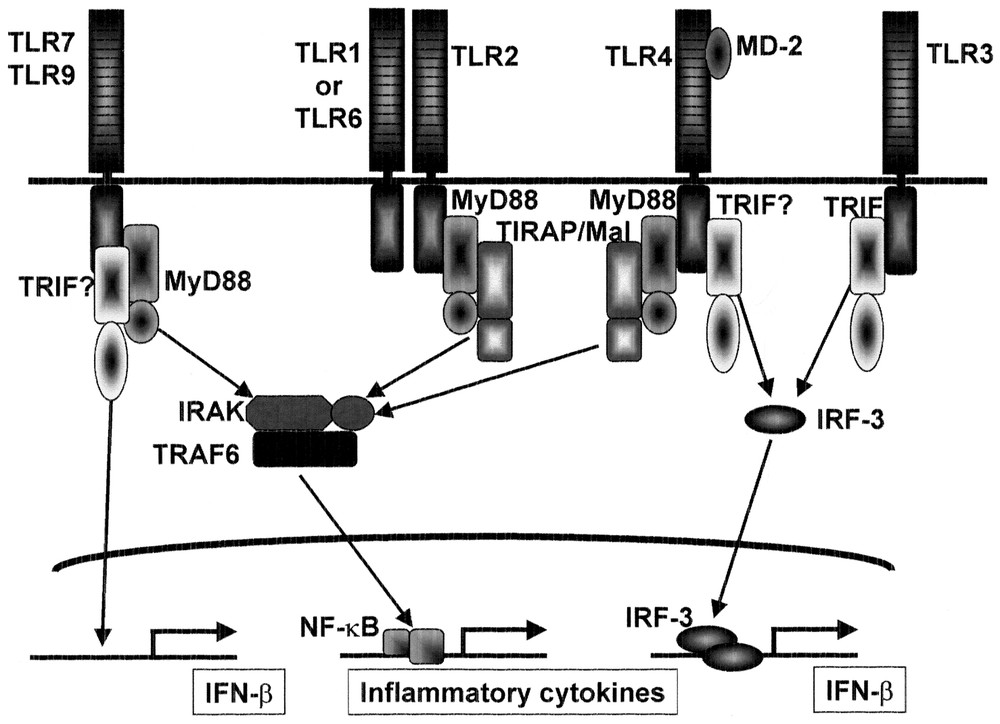
Role of adaptors in the TLR signaling. MyD88 is essential for inflammatory cytokine production in response to all TLR ligands except for TLR3 ligand. TIRAP/Mal is essential for TLR2-dependent and TLR4-dependent inflammatory cytokine production, but is not involved in the MyD88-independent pathway of TLR4 signaling. TRIF may be involved in TLR3 signaling. TRIF may also be involved in the MyD88-independent pathway of TLR4 signaling as well as interferon induction via other TLRs such as TLR7 and TLR9.
6 Self-tolerance and cross-tolerance
The exposure of macrophages to LPS induces a hypo-responsive state to a second challenge with LPS that is termed LPS tolerance [37]. LPS tolerance in macrophages/monocytes has been investigated extensively and shown to be characterized by a decline in the production of inflammatory cytokines including TNF-α, IL-6 and IL-12 and alterations to the LPS-induced activation of signaling cascades including protein kinase C, PI3K, mitogen-activated protein (MAP) kinases, and IκB kinases. However, the precise mechanism responsible for LPS tolerance remains unclear. We and others previously showed that down-regulation of the surface expression of the TLR4-MD2 complex and TLR4 caused LPS-induced LPS tolerance in mouse peritoneal macrophages and THP-1 human monocytic cell lines, respectively [38,39]. However, LPS tolerance induced by lipopeptides and LTA (cross tolerance), both of which are recognized by TLR2, has been found to occur through a distinct mechanism from LPS-induced LPS tolerance [40,41]. These findings imply that LPS tolerance was differentially induced by each TLR ligand. We analyzed the induction of LPS tolerance by several microbial components in mouse peritoneal macrophages [40,42]. LPS (TLR4) signaling is composed of the MyD88-dependent cascade that is essential for production of inflammatory cytokines and the MyD88-independent cascade that mediates the expression of IFN-inducible genes, whereas TLR2 signaling involves the MyD88-dependent cascade alone. TLR3 signaling is composed of the MyD88-independent pathway. Pre-exposure to LPS led to impaired activation of both the pathways. In contrast, pre-treatment with mycoplasmal lipopeptides (TLR2 ligand) did not affect the MyD88-independent pathway, but impaired the MyD88-dependent signaling by inhibiting LPS-mediated activation of IRAK-1. Pre-treatment with double-stranded RNA (TLR3 ligand), led to defective activation of the MyD88-independent, but not the MyD88-dependent, pathway. Imidazoquinoline compounds, TLR7 ligand, had no effect on the MyD88-independent pathway, but inhibited LPS-induced activation of MyD88-dependent signaling through down-regulation of IRAK-1 expression. Thus, cross-tolerance is induced in such a way that a given pathway, once activated, is refractory to subsequent activation, which may explain the complicated nature of LPS tolerance (Fig. 5).
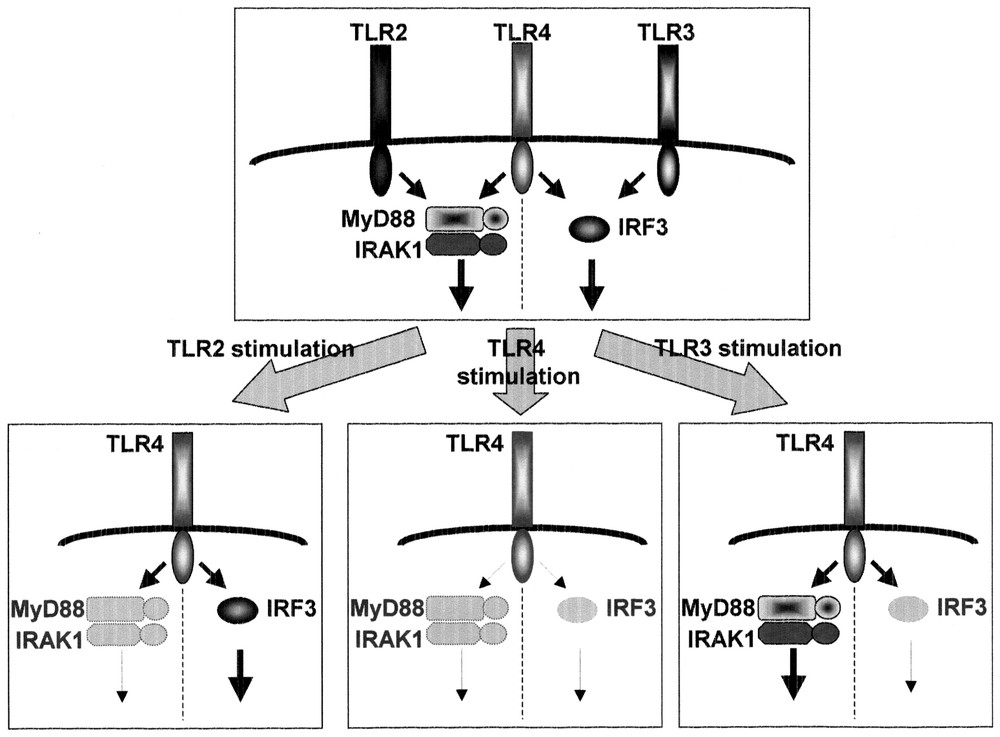
Cross-tolerance. LPS pretreatment led to the impaired activation of both the MyD88-dependent and -independent pathways. Pretreatment with MALP-2 (TLR2 ligand) or R-848 (TLR7 ligand) affected the MyD88-dependent, but not the MyD88-independent pathway. Poly (I:C) (TLR3 ligand) affected the MyD88-independent, but not the MyD88-dependent, pathway.
Acknowledgements
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology in Japan.