

1 Background
Although the receptors that are responsible for innate immune recognition were difficult to find, there is nothing fundamentally unusual about them. They do not detect ‘patterns’, as is sometimes said. Rather, like all receptors, they detect individual molecules (ligands). The molecules that are detected have defined structures, and represent components of microbes that are not easily eliminated by mutation. In those cases that have been studied, direct interaction between the ligand molecules and the innate immune sensors occurs in order to elicit a signal.
The first inroad into the identification of innate immune receptors of mammals was made in 1965, with the fortuitous observation that mice of the C3H/HeJ strain were highly resistant to the lethal effect of LPS [1]. This observation was extended through the following decades, wherein it was realized that all cellular responses to LPS, including the adjuvant effect of LPS on adaptive immune responses [2–6], were impaired by a single mutation affecting a locus that was called Lps [7]. Further, the lethal effect of LPS was seen to depend upon mononuclear phagocytic cells of the host [8]. Finally, a failure to sense LPS was associated with markedly enhanced susceptibility to infection by Gram-negative bacteria [9–12].
The Lps locus was resolved by positional cloning in 1998 [13,14]. At that time, it became clear that all LPS sensing was dependent upon a single receptor protein known as TLR4. TLR4 was one of five paralogous proteins known to exist at that time, each with homology to the Drosophila Toll protein – a molecule known to fulfill a dual role in development and in immunity. Flies with mutations in Toll had been shown to be hypersusceptible to infection by fungal pathogens [15], and in the course of time, were also shown to be hypersusceptible to infection by Gram-positive bacteria [16]. The mammalian TLRs, first identified on the basis of homology searches as early as 1994, were initially thought to have developmental functions [17,18]. However, the identity of Lps and Tlr4 proved otherwise, at least for that particular paralog, since no developmental problems had been noted in C3H/HeJ mice despite extensive study.
The discovery that Lps encoded TLR4 was a dramatic advance, for it suggested that each mammalian TLR might recognize a distinct microbial molecule, or at most, a small collection of such molecules. In the course of time, this hypothesis was proven correct. TLR2 (by itself or in conjunction with TLRs 1 or 6) serves as a sensor of bacterial lipopeptides [19], and glycans [20]; TLR3 recognizes double-stranded RNA [21]; TLR5 recognizes flagellin [22]; TLR9 recognizes unmethylated DNA [23]. The TLRs, collectively, respond to molecules produced by most microbes, and alert the host to the presence of infection. In some instances, specificity remains elusive. The natural ligand for TLR7 is still unknown, though this TLR senses small nucleotide-based drugs (imidazoquinolones) [24]. Human TLR10, which has no mouse counterpart but is closely related in structure to TLRs 1 and 6, also remains in search of a ligand.
2 The biochemistry of signal transduction: the functions of MyD88 and MAL/Tirap
The mammalian TLRs comprise a major branch of a protein superfamily (Fig. 1). Each TLR is a single-spanning type-I transmembrane protein characterized by numerous leucine-rich repeat motifs in the ectodomain, and each is endowed with a single ‘Toll/IL-1R/Resistance’ (TIR) motif that comprises the bulk of the cytoplasmic domain. The TIR motif is not represented only in TLRs, however. An ancient protein fold, the TIR motif is usually associated with an innate immune defensive function (even in plants, where it is an essential part of host resistance proteins). In mammals, TIR motifs are also present in proteins of the IL-1R/IL-18R family, which have immunoglobulin repeats in the ectodomain. Moreover, five cytoplasmic adapter proteins (MyD88, MAL, Trif, Tram, and Sarm) are known to have TIR motifs.
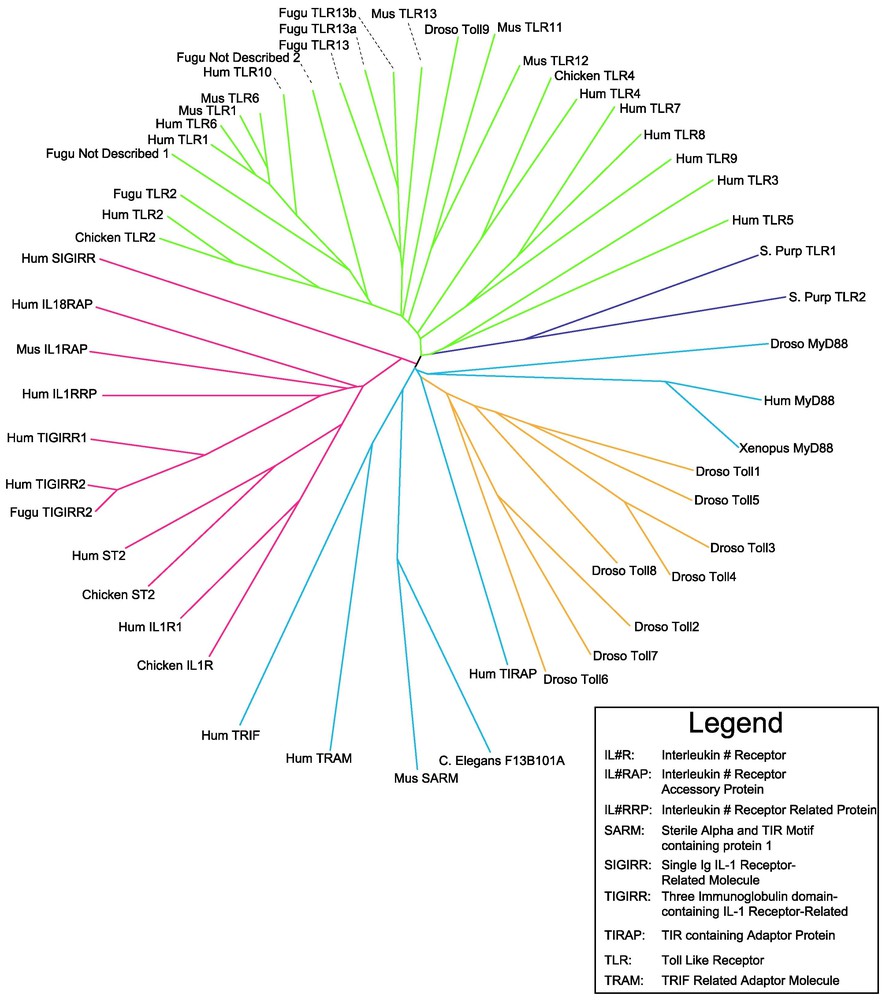
Unrooted tree of all known animal TLR paralogs. Tree was generated from a clustal W alignment of TIR domain protein sequences identified by BLASTP searches of the nr division of Genbank, and Hmmersearch of all human and mouse EST sequences translated in all reading frames. Vertebrate TLR branches are colored in green; Drosophila TLRs in orange; and representatives of the Ig-associated TIR domain receptors are colored in red. The adapter family is depicted in blue. Sea urchin TLRs are shown in purple. Note that mice have a total of twelve TLR sequences, but lack an ortholog of human TLR10. Hence, thirteen TLRs in all exist between the two species. Further, note that a single Drosophila TLR domain falls within the mammalian TLR clade, suggesting that the in the progenitor of insects and mammals, multiple TIR-domain proteins were already represented. The adapter proteins are widely scattered in the tree, indicating a particularly ancient ancestry. Interestingly, the mammalian SARM TIR is most closely related to a C. elegans TIR. Branch lengths are indicated in millions of years, using the date of divergence between birds and mammals (ca. 310 MYA) as a standard. Masquer
Unrooted tree of all known animal TLR paralogs. Tree was generated from a clustal W alignment of TIR domain protein sequences identified by BLASTP searches of the nr division of Genbank, and Hmmersearch of all human and mouse EST sequences ... Lire la suite
MyD88 was first identified as an intermediate in IL-1 receptor signaling. The IL-1 receptor had been identified as a homolog of Toll in 1990 [25], and MyD88 [26] as a related homolog in 1994 [27]. It was logical to assume that heterotypic interaction between IL-1R and MyD88 might be required for effective signaling. The case was proved with the observation that mice with targeted deletions of the MyD88 gene could not sense IL-1; neither could they sense IL-18 [28]. The role of MyD88 in LPS signaling was established subsequently [29]. At present, it appears that all TLRs except TLR3 depend upon MyD88 for signaling, at least in part.
MyD88 displays an N-terminally placed death domain that serves to recruit the interleukin receptor associated kinase (IRAK)-4, a serine kinase, which in turn phosporylates IRAK-1 and IRAK-2. IRAK-1 and IRAK-2 may serve a scaffold function, and help to recruit TRAF-6, another scaffold protein that is required for activation of numerous downstream kinases, including members of the MAP kinase superfamily and components of the signalosome complex (IKKα, β, and γ), which is responsible for phosphorylation of IκB, and consequent activation of NF-κB.
In MyD88-deficient mice, it was noted that LPS signal transduction is only partially impaired [29]. There is tardive phosphorylation of MAP kinases, and there is tardive activation of NF-κB. Moreover, some events occur without any impediment at all: for example, the phosphorylation of IRF-3, a transcription factor required for interferon-β gene expression.
The fact that residual LPS signaling activity was observed in MyD88-deficient mice prompted speculation that TLR4 must engage more than one adapter protein in order to signal. Moreover, the fact that TLR3 did not require MyD88 suggested that still other adapters might exist. A second adapter (MAL, the ‘MyD88 Adapter Like’ protein, also known as Tirap), was identified by blast searches of EST and genomic databases [30,31]. It was initially proposed that MAL/Tirap was responsible for MyD88-independent signaling [36]. However, this assertion, based upon transfection rather than germline mutations, proved to be incorrect. In 2002, the phenotype of the MAL/Tirap knockout was shown to be identical to that of the MyD88 knockout, at least with respect to signaling via TLRs 2 and 4 [32]. For TLR2, all signaling potential was lost; for TLR4, only moderate impairment of signaling was observed.
2.1 Forward genetic analysis identifies a third adapter required for TLR3 and TLR4 signaling: the Lps2 locus and Trif/Ticam-1
The success of phenotype-driven gene discovery in the identification of the mammalian TLRs as primary sensors of infection led to the use of a germline mutagen, N-ethyl-N-nitrosourea (ENU) as a tool for the production of still other innate immunodeficiency phenotypes. Hoebe and colleagues produced mice with a defect in TLR3 and TLR4 signaling caused by a single point mutation [33], mapped to mouse chromosome 17. On 1567 meioses, this mutation (in the Lps2 locus) was confined to a 216 kb interval and positionally cloned [34]. It was found to reside in the distal coding region of a third adapter protein, recently identified by homology searching and by use of the two-hybrid system, and respectively, called Trif [35], or Ticam-1 [36].
While the Trif/Ticam-1 protein was believed to be capable of inducing interferon-β gene expression through interaction with IRF-3, there was disagreement as to which TLRs it served. As has commonly been the case in the TLR field, the primacy of germline mutations over in vitro methods was demonstrated with the finding that TLRs 3 and 4 (rather than most TLRs or TLR3 alone) depended upon Trif/Ticam-1 for effective signal transduction. Hoebe and colleagues showed that the Lps2 mutation was required for effective antiviral responses, and for much of LPS toxicity in vivo [34]. They also demonstrated that the protein was an integral component of the TLR(3,4) → IRF-3 → interferon-β signaling axis [34]. Trif/Ticam-1 was also shown to mediate all of MyD88-independent signaling in that mice with mutations in both the Trif/Ticam-1 gene and the MyD88 gene showed no residual LPS responses at all [34] (Fig. 2).
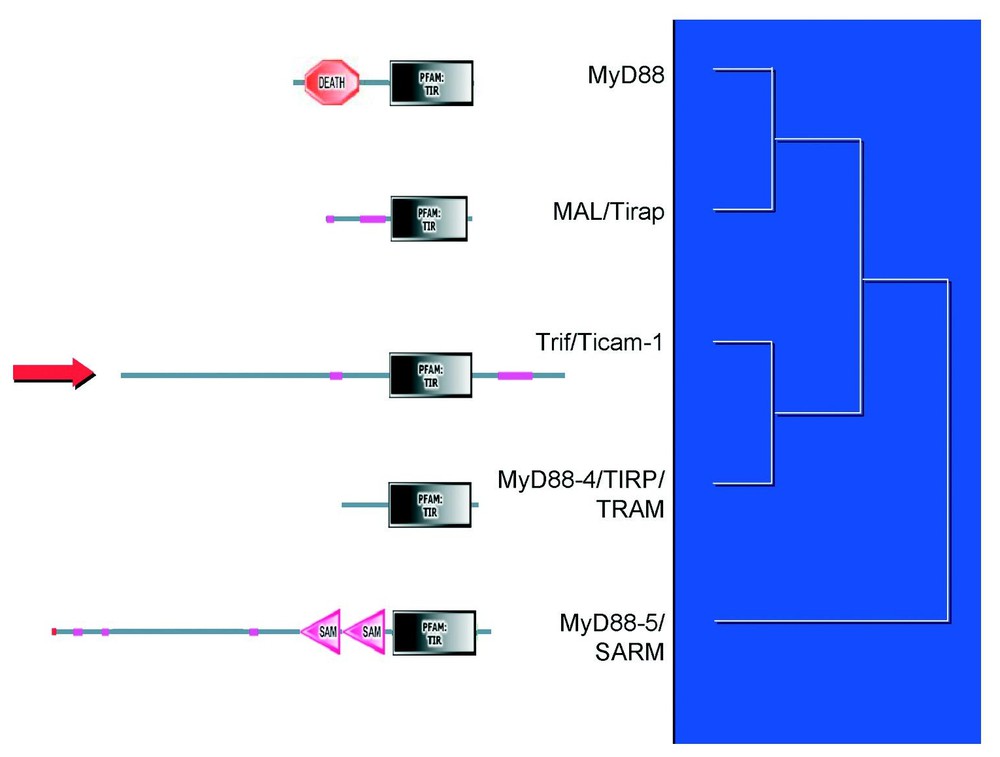
The structure and phylogeny of the TIR adapters. Dendrogram shows only phyletic relationships, and is not drawn to scale. Motifs were identified by SMART (Simple modular architecture tool).
The phenotype of homozygosity for the codominant Lps2 allele was subsequently shown to be similar or identical to that of homozygosity for a Trif/Ticam-1 knockout allele [37]. However, an additional observation was made using Lps2 homozygous mice that pointed to the existence of still another adapter, required for LPS signaling.
2.1.1 Trif/Ticam-1 independent cells
FACS analysis of TNF production in cells from wild type mice showed that virtually all peritoneal macrophages respond to LPS by producing TNF protein. On the other hand, no cells from MyD88-deficient mice produce TNF protein in any great quantity. Remarkably, cells from Lps2 mutant homozygotes are of two types: some produce TNF in response to LPS and others do not [34]. The cells that show residual responsiveness are termed ‘Trif-independent’. Trif independence cannot be attributable to MyD88 signaling, since MyD88 is represented in all cells, and if it were responsible for the ‘rescue’ that is observed, rescue should be uniform (and not bimodal). Hence, the existence of an ‘adapter X’ was posited, and further, it was suggested that adapter X was most likely identical to Tram, the TIR adapter protein that is the closest phylogenetic relative of Trif [34]. Early indications from the knockout of Tram suggest that this hypothesis is correct [38]. Tram functions as a component of the MyD88-dependent pathway downstream from TLR4, and in certain cells, can partly replace the function of Trif (Fig. 3).
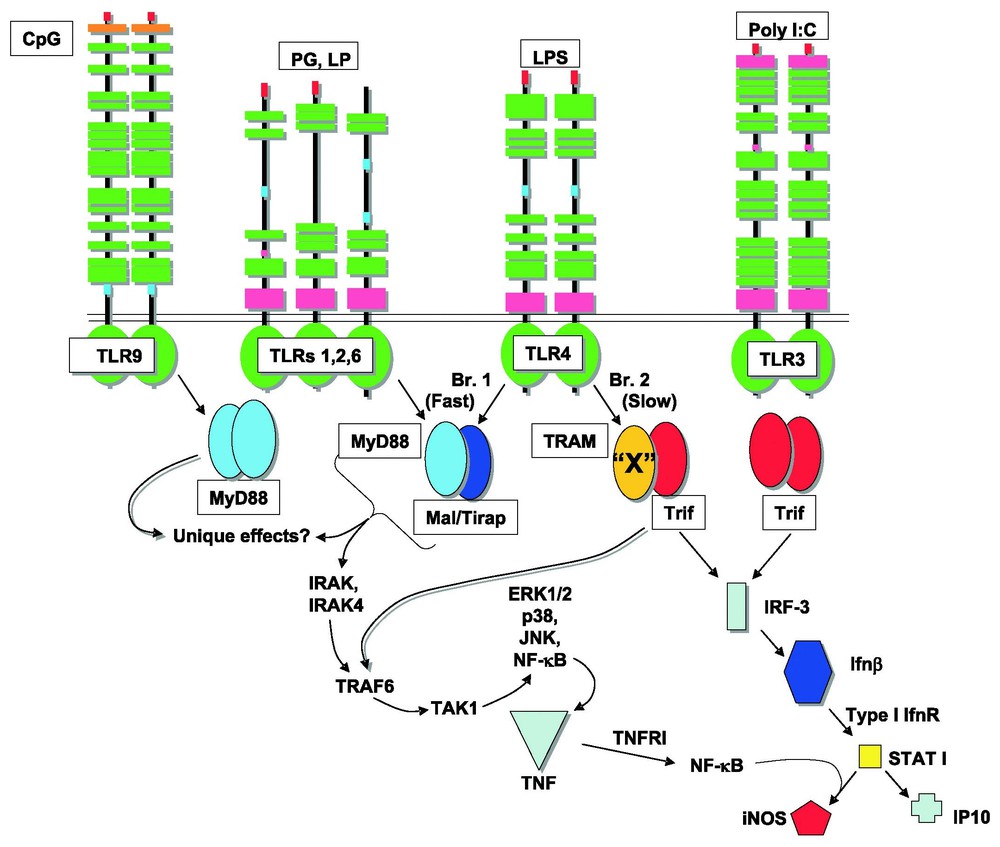
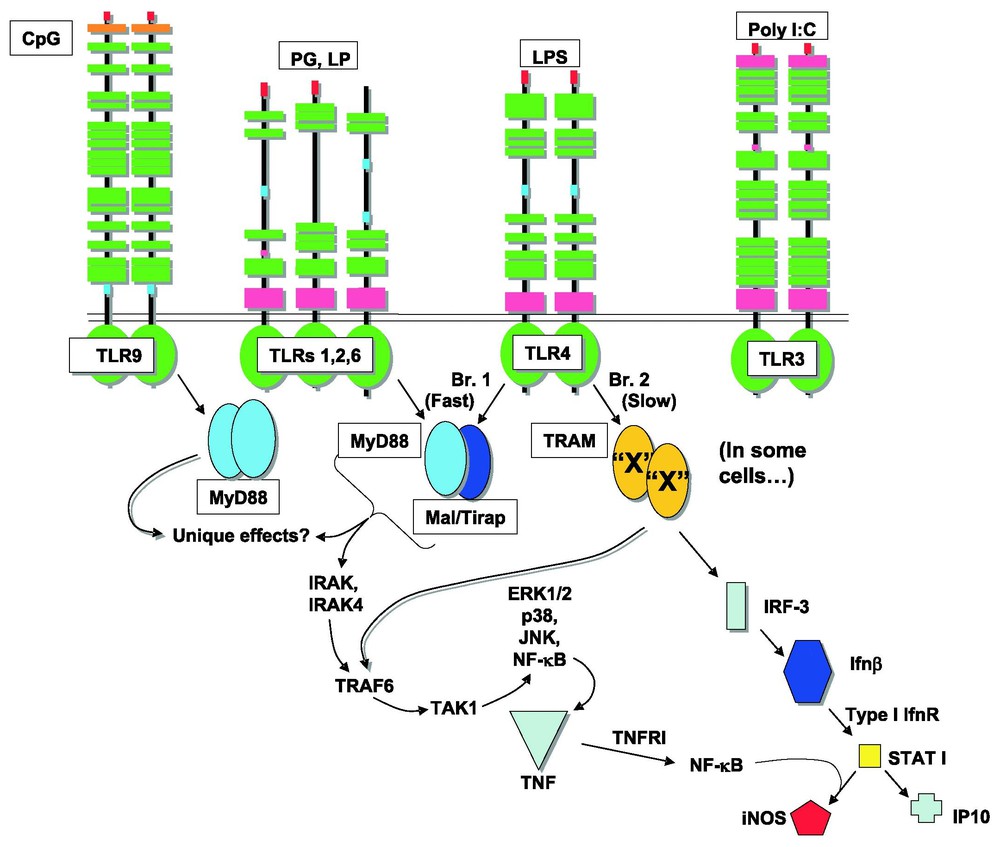
TLR signaling pathways as presently understood. A. The postulated dimeric structure of adapter proteins, and the downstream events that they initiate. Four adapters serve the LPS receptor, TLR4. Two adapters serve TLR2, and one adapter serves TLR3; a different adapter serves TLR9. ‘X’, an adapter known to exist on the basis of studies in TrifLps2 mice, is believed to be equivalent to Tram. B. In the absence of Trif, Tram homodimers may form in some (but not all) macrophages, and can serve MyD88-independent signaling events. Masquer
TLR signaling pathways as presently understood. A. The postulated dimeric structure of adapter proteins, and the downstream events that they initiate. Four adapters serve the LPS receptor, TLR4. Two adapters serve TLR2, and one adapter serves TLR3; a different adapter ... Lire la suite
Tram appears to cooperate with Trif in all of Trif's LPS responses, including the upregulation of costimulatory proteins. But plays no part in the upregulation of costimulatory proteins initiated at the level of TLR3 [38]. A plausible model of TIR adapter function would hold that all of the adapters function as homodimers or heterodimers, much as all of the TIR-domain receptors are believed to function, as depicted in Fig. 3.
2.2 Sarm
The fifth known TIR adapter protein is also endowed with Sterile Alpha Motif (SAM) domains, and at present, its function is entirely unknown. This is the most distant of the TIR adapters, displaced from the other members of the family by a great evolutionary distance. The Sarm TIR motif is most similar to a TIR motif observed in C. elegans. It is not clear whether Sarm is actually required for TLR signal transduction or not. Either gene knockout or forward genetic methods may soon provide an answer.
3 Which TIR adapters serve the adaptive immune response?
The adjuvant effect of individual microbial molecules has been known for a very long time. By 1955, LPS was shown to be an adjuvant for adaptive immune responses [39], and as already noted, the Lps locus was shown in 1975 to be required for this biological endpoint of LPS action [3]. Hence adjuvanticity depends upon TLR4 [14], and a biochemical pathway for this effect would seem definable.
Adjuvanticity is dependent in large part upon the upregulation of costimulatory proteins (e.g., CD80, CD86, and CD40) that engage receptors on T cells and coordinate the mitogenic response to a specific antigen. LPS upregulates these costimulatory proteins on antigen-presenting cell, and do so by engagement of TLR4. However, the early and oft-repeated supposition that TLR-induced NF-κB activation was responsible for upregulation [40] proved to be incorrect. In MyD88-deficient cells, upregulation proceeds unimpeded [41,42]. On the contrary, in Trif-mutant cells, despite persistent activation of NF-κB, upregulation of costimulatory proteins by LPS is abolished [43].
Recently, Hoebe et al. have demonstrated that LPS-induced upregulation of costimulatory proteins proceeds directly through the TLR4 → Trif → TBK1 → IRF-3 → IFNb axis, and depends upon activation of the type I interferon receptor [43]. On the other hand, upregulation of costimulatory proteins may be achieved through two alternative pathways when the inducer is dsRNA. One pathway is dependent upon TLR3, Trif and its downstream signaling partners. The other pathway is obscure, but is TLR3- and Trif-independent (Fig. 4). A quantitative trait locus on chromosome 7 (designated dsRNA1) defines the alternative pathway, and is presently being mapped to high resolution.
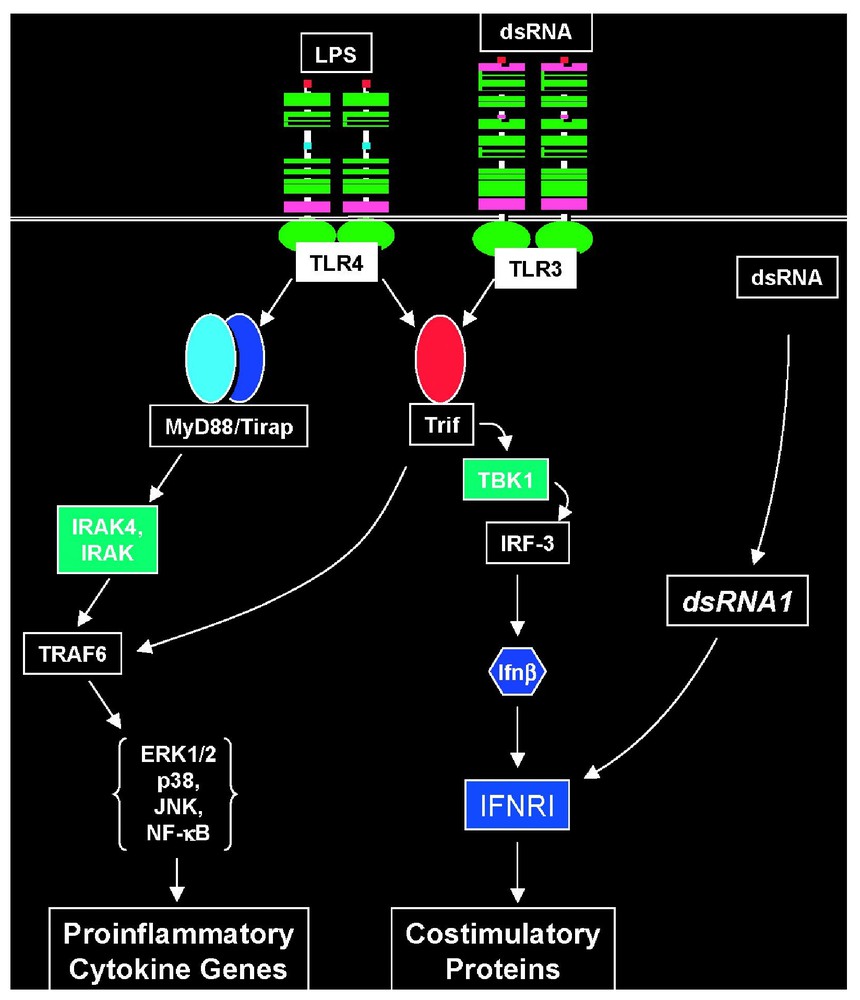
Pathways to inflammatory and adjuvant responses. TLRs 3 and 4 both signal the production of interferon-β by way of Trif, and type I interferon is absolutely required for upregulation of costimulatory molecules on antigen presenting cells: a prerequisite for adjuvanticity. However, dsRNA also induces upregulation of costimulatory molecules via a second pathway, independent of both TLR3 and Trif. This second pathway is genetically defined by the dsRNA1 locus, mapped to chromosome 7.
4 The global importance of TLR signaling: which TLRs are required for management of which pathogens?
TLR4 is required for effective containment of Gram-negative infections, as previously mentioned [10,11]. TLR2 contributes to the containment of at least some Gram-positive bacterial infections [44]. Trif, and by implication TLR3, is known to be required for effective management of infection by MCMV [34], and TLR9 has been shown to be important in the containment of Herpes simplex virus infection [45]. Unpublished data (Tabeta, et al., submitted) indicate that the TLR9/MyD88 couple, and the TLR3/Trif couple, are both of key importance for containment of MCMV infection in vivo, and further, have shown that both signaling pathways are important for effective production of type I interferons.
Hence, a distinct division of labor is apparent, but also, it is evident that many of the same proteins that defend the host against viral infection also protect against bacterial infection (witness the sharing of Trif by TLRs 3 and 4).
4.1 Blockade of TLR signaling: the therapeutic opportunies
It is possible to block TLR4 signal transduction selectively and specifically using small molecular antagonists [46]. In years to come, antagonists aimed at the interruption of TIR domain interactions [47], and at the kinases that are immediately linked to TIR domain proteins (IRAK-4, TBK-1) may be used to create a highly specific form of immunosuppression.
As previously noted, the TLRs evolved to sense infection in a timely fashion and prevent small inocula of microbes from progressing to cause overwhelming infection. At the same time, the TLRs deliver the lethal signal that overwhelming infection creates. Without TLR4, LPS is a harmless substance; with TLR4, LPS can readily kill the mammalian host. Is it wise to block TLR signaling? Or is it better to maintain signaling and somehow ‘ride out the storm’? Or does the best course lie somewhere in between? Or does it depend upon circumstance? New therapeutic opportunities have been created by our newfound understanding of innate immune sensing, but precise knowledge of the most prudent therapeutic approach is still a bit beyond our grasp. In all likelihood, the decision to block TLR signaling may depend upon the stage of infection, the pathogen involved, and the efficacy of antibiotic therapy in dealing with that pathogen.
5 Do TLRs support immune responses in which there is no exogenous signal?
By their very nature, TLRs ought not to recognize endogenous molecules of the host. An absence of such interactions is the sole mechanism for self tolerance exercised by this family of germline-encoded receptors. On the other hand, sterile inflammation is an authentic phenomenon, and it must be initiated by receptors of a kind. Might the TLRs be involved? The question arises in several different settings, and while it is likely that many of the purported ‘endogenous’ ligands for TLRs will ultimately prove to be artifactual, one cannot categorically exclude the possibility that a small number of true endogenous activators might exist.
If it is accepted that an adjuvant is absolutely required for an adaptive immune response, it might be argued that TLRs are required for events such as graft rejection to occur, despite the absence of any microbial presence. Is this the case? Or are TLRs merely a facilitator rather than a sine qua non for adaptive immune responses?
Suggestive evidence indicates that TLRs may play some role in graft rejection [48], although the possible contribution of microbial flora to the process of rejection has not been excluded. One might point to truly sterile grafts, such as blood transfusions, and ask whether failure to develop stable chimerism is TLR dependent. To date, however, the question has not been addressed.
In a similar manner, it is broadly suspected that TLRs might play some role in autoimmune processes. It is known that TNF makes an important contribution to autoimmune injury in selected diseases (Crohn's disease, rheumatoid arthritis, and ankylosing spondylitis, for example). Moreover, there are only a limited number of ways in which TNF biosynthesis can be induced: all pathways must converge on the activation of NF-κB, and upon elimination of the translational blockade that normally keep TNF production in check. Do autoimmune diseases involve dysregulation of TLR signaling pathways? The question is one that will be investigated widely in the near term.
Vous devez vous connecter pour continuer.
S'authentifier