

1 Introduction
Described for the first time by James Parkinson in 1817 in his Essay on the Shaking Palsy, Parkinson's disease (PD) is the second most common age-related, progressive neurodegenerative disorder after Alzheimer's disease. With a mean age at onset of 55, PD affects nearly 2% of the population over age 65. Clinically, it is characterized by a triad of invalidating neurological symptoms – bradykinesia, rigidity and resting tremor – that become manifest when more than 60% of the melanized dopaminergic neurons of the substantia nigra pars compacta (SNc) have degenerated [1–3]. Lewy bodies (LBs), ubiquitylated intraneuronal protein inclusions enriched in α-synuclein, are a neuropathological hallmark invariably associated with idiopathic PD [4,5]. Although the cause of this disease remains obscure, there is evidence that both environmental risk factors and genetic predisposition can contribute to its development.
2 Sporadic Parkinson's disease: an environmental disorder?
In 1983, the discovery of a severe and unremitting parkinsonian syndrome in young drug-addicts involuntary intoxicated with the synthetic narcotic contaminant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) provided support for the hypothesis that exposure to environmental neurotoxins can lead to PD [6]. In experimental models of parkinsonism, the active MPTP metabolite, 1-methyl-4-phenyl pyridium (MPP+), was shown to be taken up specifically in monoaminergic neurons via the dopamine, norepinephrine, and serotonin transporters, and to inhibit the multienzyme complex I of the mitochondrial electron-transport chain [7–9]. The degenerative changes caused by MPP+ are most prominent in dopaminergic neurons, suggesting that these cells are particularly vulnerable to mitochondrial complex I impairment [9]. Since the discovery of MPTP, epidemiological investigations have suggested that exposure to pesticides might be associated with a high risk of developing PD [10]. Experimental studies support this possibility. Like MPP+, the structurally related herbicide, paraquat (1,1′-dimethyl-4,4′-5 bipyridinium), leads to selective degeneration of nigral dopaminergic neurons when administered to mice [11]. Similarly, the common insecticide and fish poison, rotenone, delivered to the rat intravenously or subcutaneously, was reported to induce an apparently selective death of the nigrostriatal dopaminergic neurons, despite the widespread inhibition of mitochondrial complex I activity that it causes [12,13]. More recent investigations demonstrated that the deleterious effects of rotenone and annonacin, another natural lipophilic mitochondrial complex I inhibitor, also affect striatal cell populations in the rat according to a pattern of degeneration compatible with that observed in atypical parkinsonian syndromes [14,15]. Thus, although the nigral dopaminergic neurons are probably not selectively vulnerable to complex-I impairment, some forms of parkinsonism might be induced by environmental toxins that affect the activity of this complex. The finding of a complex-I deficiency in the SNc and the platelets of PD patients is not only consistent with this notion, it also provides a first direct link between idiopathic PD and experimental models of parkinsonism [16]. A second major link is provided by the finding that MPTP, paraquat and rotenone are capable of inducing the formation of intracellular fibrillar protein inclusions immunoreactive to α-synuclein and reminiscent of LBs (Fig. 1) [12,17,18].
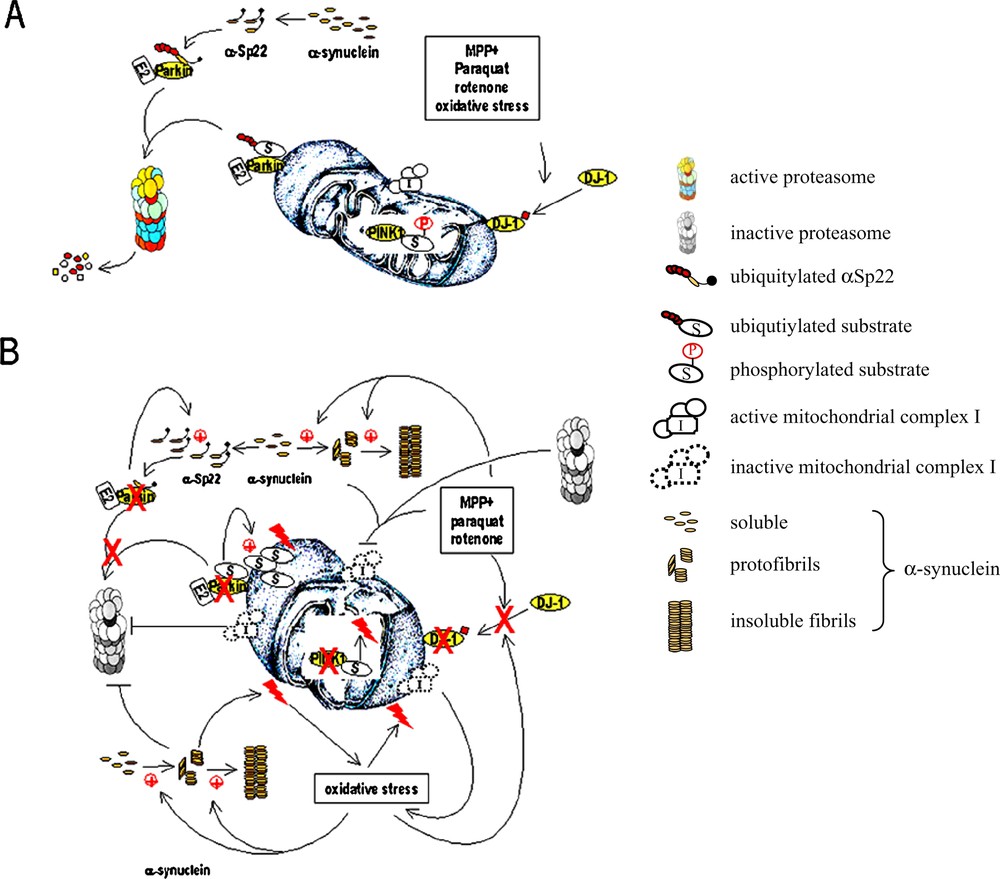
Pathways regulating the integrity of mitochondria that are affected in Parkinson's disease. (A) In healthy individuals, the products of three genes involved in familial parkinsonian syndromes protect nigral dopaminergic neurons against mitochondrial damage. Parkin controls mitochondrial homeostasis by regulating the turnover of one or more as yet unknown substrates located on the outer mitochondrial membrane. It also helps maintaining normal α-synuclein levels by ubiquitylating the O-glycosylated α-synuclein isoform αSp22. Oxidative or environmental stress induces the conversion of DJ-1 to a more acidic isoform which is translocated to the outer mitochondrial membrane where it exerts its protective effects. The inner mitochondrial protein PINK1 regulates the function of mitochondria by phosphorylating specific substrates to be identified. (B) In PD, one or more of these neuroprotective mechanisms are compromised. Partial or complete loss of Parkin function results in the aberrant accumulation of its mitochondrial substrates (⊕), thus sensitizing cells to mitochondrial damage. The abnormal increase in α-synuclein levels (⊕) leads to the formation of protofibrils and insoluble fibrils, followed by proteasomal impairment, a process that cannot be prevented in the absence of functional Parkin. Accumulation of α-synuclein also causes direct mitochondrial damage and, in synergy with environmental toxins, dysfunction of the multienzyme complex I of the mitochondrial electron transport chain. The resulting depletion of adenosine triphosphate amplifies proteasomal impairment, which, in turn, exacerbates the effect of mitochondrial complex I inhibitors and aggregation of α-synuclein. The toxicity of complex-I inhibitors and the resulting oxidative stress cannot be counteracted if DJ-1 fails to translocate to the outer mitochondrial membrane or if it is inactive. Oxidative stress further favours the aggregation of α-synuclein, proteasomal damage, complex I impairment; a cascade of events leading to loss of the mitochondrial transmembrane potential, mitochondrial swelling, rupture of the outer mitochondrial membrane and the release of proapoptotic molecules.
3 α-Synuclein: the culprit in Parkinson's disease?
α-Synuclein has been suspected to play a central role in the pathophysiology of PD since 1997, when the A53T mutation was identified in the large Italian–American Contursi kindred and in three unrelated kindreds of Greek origin as the first causative gene alteration responsible for an autosomal-dominantly-inherited parkinsonian syndrome [19]. A second (A30P) and, more recently a third missense mutation (E46K) were subsequently identified in the α-synuclein gene [20,21]. These point mutations, which account for only a small number of cases with autosomal dominant PD, are associated with a clinical phenotype similar to idiopathic PD, although with an earlier age at onset, a more rapid disease progression and atypical clinical signs. Elevated levels of normal α-synuclein might be a more frequent cause of disease, as suggested by the discovery of duplications and triplications of a genomic region including the α-synuclein gene and leading to autosomal dominant PD in the absence of any other α-synuclein gene mutation [22–24,118]. Remarkably, the severity of the parkinsonian phenotype appears to correlate with the number of supernumerary copies of the α-synuclein gene, three of which cause a phenotype indistinguishable from idiopathic PD, whereas four copies lead to Parkinsonism with severe disease progression and atypical clinical features. Although these findings should be regarded with caution – the reported genomic alterations involve a region containing 17 additional genes that could also be implicated in the disease – they imply that impaired metabolism of normal α-synuclein, known to accumulate massively in LBs, could be the primary cause of neurodegeneration in idiopathic PD. Furthermore, this discovery provides an explanation for the intriguing observation that the over-expression of normal human α-synuclein in Drosophila and rodents by means of transgenesis or viral vector-mediated gene delivery can lead to dopaminergic neurodegeneration [25–29].
The reasons why normal or altered α-synuclein are selectively toxic for nigral dopaminergic neurons are probably diverse, involving both a loss of function by α-synuclein sequestered in protein aggregates and a toxic gain of function by oligomerized and/or fibrillar α-synuclein. α-Synuclein has been reported to negatively regulate the activity of tyrosine hydroxylase by maintaining the protein in the dephosphorylated inactive form, thus modulating dopamine biosynthesis in catecholaminergic neurons [30]. In addition, α-synuclein was proposed to negatively regulate the function of the dopamine transporter in dopaminergic nerve terminals by controlling the amount of the transporter at the plasma membrane [31]. Alteration of α-synuclein by disease-causing mutations, its oligomerisation or its sequestration in LBs in PD might reduce the amount of soluble functional protein available, leading to the partial loss of the above-mentioned modulatory effects and to the abnormal increase in intracellular synaptic dopamine concentrations. In the absence of compensatory mechanisms allowing efficient uptake and storage of excessive dopamine in synaptic vesicles, these changes could create favourable conditions for the production of deleterious reactive oxygen species generated by the intraneuronal metabolism and the auto-oxidation of dopamine [32].
The currently available mouse models characterized by the overproduction of normal or mutated α-synuclein do not fully recapitulate the clinical and neuropathological features of PD. Nevertheless, they have provided evidence that the abnormal aggregation of α-synuclein can be associated with neuronal demise and/or degeneration [29,33–35]. Inclusions in these mice are not always fibrillar in nature, which supports recent models proposing that soluble, protofibrillar α-synuclein might be a major toxic species in PD: soluble normal and mutated α-synuclein can predispose neuronal cells to death in the absence of neuronal inclusions [36,37]; at least two of the three identified α-synuclein gene mutations promote the formation of protofibrils of α-synuclein [38]; protofibrils are stabilized by dopamine–quinone adducts that prevent their conversion into insoluble amyloid fibrils [39]; in vitro studies have shown that protofibrillar α-synuclein is capable of permeabilizing cellular membranes by forming pore-like structures like some bacterial toxins [40]. Such pores could affect the membranes of synaptic vesicles and disrupt the vesicular storage of dopamine. They could also damage mitochondria, leading to dissipation of the transmembrane potential and ultimately to the release of proapoptotic proteins. Although the relevance of these findings needs to be demonstrated in cellular and animal models, there is evidence that the over-expression of α-synuclein impairs mitochondrial function and promotes oxidative stress (Fig. 1) [41,42]. There is also accumulating evidence that links overproduction of α-synuclein to increased susceptibility to mitochondrial toxins. Accordingly, whereas α-synuclein-deficient mice are resistant to MPTP toxicity, the overproduction of α-synuclein appears to sensitize transgenic mice to the deleterious effects of MPTP, leading to the selective enhancement of mitochondrial pathology in the SN and the striatum (Fig. 1) [43].
4 Parkin, α-synuclein and the ubiquitin-proteasome pathway: pieces of a puzzle?
A putative mitochondrial action was also recently attributed to Parkin, the product of the second gene identified in 1998 as unambiguously related to a monogenic parkinsonian syndrome: autosomal recessive juvenile Parkinsonism (ARJP). Parkin was reported to be associated with different subcellular compartments – Golgi network, endoplasmic reticulum, synaptic vesicles, postsynaptic densities – and is also localized on the outer mitochondrial membrane in the mouse brain and in PC12 cells genetically modified to produce the human protein [44–47]. A mitochondrial location could explain the protective effect of Parkin in a cell-death paradigm mediated by ceramide, in which Parkin delayed mitochondrial swelling, rupture of the outer mitochondrial membrane, the release of cytochrome c and activation of caspase 3 [46]. In support of a central role of Parkin in maintaining mitochondrial homeostasis, Parkin-deficient Drosophila exhibited adult-onset muscle degeneration preceded by early and severe mitochondrial pathology with mitochondrial swelling and reduced number of cristae [48]. Although mitochondria were intact at the ultrastructural level in a mouse model in which the parkin gene was invalidated by homologous recombination, a differential proteomic analysis demonstrated that several components of the mitochondrial respiratory chain were down-regulated in these mice compared to wild type littermates [49,50]. Importantly, these alterations were associated with decreased respiratory capacity in mitochondria isolated from the striatum of Parkin-deficient mice [50].
Parkin is by far the protein the most frequently involved in familial PD. Since the gene was first identified, many different combinations of point mutations, exon deletions and multiplications have been found to be responsible for nearly 50% of familial cases with autosomal recessive inheritance and age at onset before 45, and more than 15% of isolated early-onset cases [51–54]. These mutations result in a broad clinical spectrum, including phenotypes resembling levodopa-responsive dystonia and idiopathic PD, with ages at onset up to 74 [51,55,56]. Surprisingly, in all but one of the few cases examined neuropathologically, LBs were absent [57–61]. Loss of Parkin function is thought to be the mechanism involved in the disease, although there is evidence that some mutations preserve at least partially its ubiquitin-protein ligase activity and that others could act dominantly [51,56,61,62]. In contrast to the function of α-synuclein, which is still enigmatic, the function of Parkin was elucidated rapidly after the discovery of its gene. Parkin is an E3 ubiquitin-protein ligase that participates, in conjunction with specific E2 conjugating enzymes, in the selective ubiquitylation of a subset of protein substrates, leading to their degradation by the ubiquitin-proteasome proteolytic pathway [63–65]. Loss of this function was proposed to lead to the aberrant accumulation of potentially toxic substrates and consequently to neurodegeneration. In agreement with the multiple subcellular locations of Parkin, the Parkin substrates identified thus far have putative or ascertained roles in cell processes as diverse as cell signalling (PaelR), cell cycle control (cyclin E), protein biosynthesis (the p38 scaffold subunit of the multi-aminoacyl-tRNA synthetase complex), cytoskeletal dynamics (α/β-tubulin), vesicular and synaptic functions (CDCrel-1, synaptotagmin IX, synphilin, O-glycosylated α-synuclein) [64,66–74]. In general, these substrates appear to be aggregation-prone proteins that promote cell death upon over-expression in cells and/or in Drosophila and rodent models, a process that, in some cases, was reported to be prevented by Parkin [66,67,69,75–77]. Remarkably, the levels of some of these substrates were shown to be increased in brains of patients with parkin-related PD [66,72–74].
Which, if any, of these multiple substrates are involved in the selective degeneration of the dopaminergic neurons of the SNc in PD caused by parkin-gene mutations is still unclear. According to one of the most tempting current hypotheses, like α-synuclein-related and sporadic PD, PD due to parkin gene mutations might be an α-synucleinopathy. This hypothesis was fueled by the discovery of an enzyme-substrate relationship between Parkin and an O-glycosylated isoform of α-synuclein present in human, but not rodent brain [72]. Although the existence of this minor α-synuclein species is debated, other studies suggest that Parkin protects from the deleterious effects of α-synuclein in primary neuronal cultures, in Drosophila and in rodents [37,77,78,117]. In particular, it has been proposed that overproduction of α-synuclein disrupts proteasomal activity and that proteasomal function can be rescued by Parkin (Fig. 1) [37]. The mechanisms by which an E3 ubiquitin-protein ligase acting upstream of the proteasome could protect against proteasomal dysfunction are, however, obscure. Mutant Parkin may also be indirectly linked to α-synuclein function through the control of the turnover of the α-synuclein partner synphilin [71]. The functional interplay between Parkin and α-synuclein might take place at the synapse, where these proteins seem to have partially opposing actions. In an α-synuclein-deficient transgenic mouse line, striatal dopamine levels were significantly decreased, whereas they tended to increase in Parkin-deficient mice [49,79,80]. Parkin-deficiency was also associated with reduced levels of dopamine uptake in embryonic mesencephalic neurons, an effect that was reproduced in cells over-expressing α-synuclein [31,80]. Finally, the levels of the dopamine and vesicular monoamine transporters were decreased in Parkin-deficient mice and in a human dopaminergic cell line over-expressing the pathogenic A53T α-synuclein variant [80,81]. Parkin and α-synuclein may therefore play essential and antagonistic roles in the modulation of vesicular neurotransmitter storage and release in dopaminergic synapses, although their effects are probably not restricted to this neurotransmitter system.
Parkin has also been proposed to participate, with α-synuclein, in the biogenesis of LBs, as suggested by (i) the absence of this neuropathological hallmark in most of the examined cases of parkin-related PD, (ii) the tendency of normal Parkin and a number pathogenic variants to misfold and/or accumulate in aggresome-like structures, (iii) the presence of Parkin immunoreactive material in LBs [57–60,82–88,119]. Although other groups failed to confirm the latter finding, Parkin was shown to contribute, with α-synuclein and synphilin, to the formation of ubiquitylated Lewy body-like inclusions in transfected cells [71,89]. Furthermore, there is evidence that Parkin promotes the aggregation of its substrates in vitro, and immunoreactivity corresponding to several of these proteins has been found in LBs in sporadic PD [67,90–93]. Therefore, like α-synuclein, Parkin might not only be the cause of the disease in cases with parkin gene mutations, but also play a more general role in the pathogenesis of idiopathic PD. Oxidation or nitrosylation of the protein and/or its sequestration in LBs are mechanisms that could lead to partial loss of Parkin function in the absence of parkin gene mutations. An aberrant S-nitrosylated, inactive isoform of Parkin was recently found in the brains of patients with sporadic PD, but not Alzheimer's disease [94]. Remarkably, this modification of Parkin was also observed in mice intoxicated with MPTP, suggesting that it could be generated following inhibition of mitochondrial complex I.
Besides Parkin, another component of the ubiquitin-proteasome pathway, the ubiquitin carboxyterminal hydrolase L1 (UCH-L1), was suspected to cause an autosomal dominant form of PD in two affected siblings of a single kindred [95]. The I93M variant of UCH-L1, an enzyme thought to play an essential role in recycling the ubiquitin monomers required for protein ubiquitylation, was shown to affect the catalytic activity of the protein in vitro. Furthermore, the absence of a functional UCH-L1 protein in gad mice results in dysfunction of the ubiquitin-proteasome pathway, associated with protein accumulation and axonal pathology affecting primary sensory axons in the gracile nucleus of the medulla [96]. Recent studies have attributed a previously unrecognized ubiquitin-protein ligase activity to UCHL-1, which is partially impaired by the I93M mutations, whereas it is enhanced by the S18Y polymorphism, reported to confer protection against the development of PD [97]. However, due to the absence of UCH-L1 gene mutations in the other sporadic and familial PD cases examined thus far, it remains uncertain whether this protein is really involved in the pathogenesis of PD.
There is also evidence that the ubiquitin-proteasome pathway is affected selectively in the SN of sporadic PD cases, in which both the proteolytic activities and the subunit composition of the proteasome were reported to be altered [98,99]. Importantly, there seems to be a functional relationship between proteasomal impairment and deficits in mitochondrial complex-I activity. In a recent study, inhibition of the proteasome in primary cultures of rat midbrain neurons was shown to exacerbate the toxicity of the complex-I inhibitors rotenone and MPP+. These agents themselves caused proteasomal dysfunction by depleting cellular adenosine triphosphate (Fig. 1) [100]. Thus, failure of the ubiquitin-proteasome system could be a pathogenic pathway shared by familial parkinsonian syndromes and sporadic PD.
5 Mitochondria: the clue to the pathogenesis of familial and sporadic Parkinson's disease?
Three additional genes have been implicated in familial parkinsonian syndromes: they encode the nuclear receptor, Nur-related factor 1 (NR4A2), DJ-1 and PTEN-induced kinase 1 (PINK1) [101–103]. The contribution of NR4A2 gene mutations to the pathogenesis of PD is certainly minor. Indeed, despite the report of two heterozygous non-coding exon 1 mutations (, ) in 10 out of 107 European cases with PD and a positive family history, neither of these mutations nor any other NR4A2 mutations were detected in 202 additional cases with apparently autosomal dominant PD [104,105]. The involvement of Nur-related factor 1 in the differentiation and maintenance of the nigral dopaminergic neurons is nevertheless of interest [106]. The identified non-coding NR4A2 mutations were shown to cause a significant decrease in NR4A2 mRNA levels in transfected cells. The homozygous disruption of this gene in mice prevents the generation of midbrain dopaminergic neurons and brain dopamine content is reduced in heterozygous animals [107].
DJ-1 and PINK1 will probably turn out to be more informative with respect to the mechanisms underlying the pathogenesis of PD, although their biological roles and their links to dopaminergic neuronal degeneration are far from being elucidated. Several DJ-1 and two (G309D, W437OPA) PINK1 gene mutations were recently identified in early-onset PD cases with autosomal recessive inheritance [108–110]. Although mutations in these genes will probably be less frequently involved in parkinsonian syndromes than parkin gene mutations, their actual prevalence remains to be established. According to recent studies, DJ-1 plays multiple roles in cell transformation, fertility, RNA stabilization, androgen receptor signalling and, importantly, the oxidative stress response [111]. DJ-1 undergoes a shift to an acidic isoform in the presence of reactive oxygen species and has protective antioxidant activity in cells exposed to oxidative stress (Fig. 1) [112–114]. However, oxidative stress is one of several ‘toxic paradigms’ in which DJ-1 was reported to be protective, leading to the concept that, like Parkin, DJ-1 might be a ‘multipurpose neuroprotective agent’ [114,115]. Remarkably, treatment of cells overexpressing DJ-1 with the inhibitors of mitochondrial complex I, paraquat and MPP+, was reported to induce a massive relocalization of DJ-1 – usually found in the cytoplasm and the nucleus – to the outer mitochondrial membrane [116]. This relocalization was strictly dependent on the oxidative conversion of a cysteine at position 106 of the DJ-1 protein to a cysteine–sulphinic acid; it was also a prerequisite for the protective activity of DJ-1, suggesting that, like Parkin, DJ-1 might play a key protective role against mitochondrial damage (Fig. 1) [116]. An incentive for the revival of the ‘mitochondrial hypothesis’ in PD was also provided by the discovery of the first PD gene encoding a mitochondrial protein: PINK1 [103]. PINK1 is a putative kinase predicted to be synthesized as a precursor with an N-terminal mitochondrial targeting signal. It contains a domain homologous to Ca2+/calmodulin-dependent serine/threonine kinases, which is affected by the two-point mutations identified so far. Preliminary studies indicate that wild-type PINK1, but not a pathogenic variant, preserves the mitochondrial membrane potential in conditions known to compromise mitochondrial integrity – i.e. treatment with the proteasomal inhibitor MG132 – thus protecting cells against apoptotic cell death (Fig. 1) [103].
In conclusion, mitochondria are emerging as a leitmotif uniting the disparate genetic and environmental causes that appear to underlie the development of a parkinsonian phenotype (Fig. 1). Future studies will help clarify whether and how the diverse proteins linked to familial PD and the environmental toxins suspected to intervene in sporadic PD fit into a common biochemical pathway centred around mitochondria and their functional integrity.