

1 Introduction
The worldwide and continuous extension of human lifespan has met with a concomitant increase in the incidences of age-related diseases such as Alzheimer's disease (AD) or Parkinson's disease (PD). This rise in neurodegenerative diseases makes modern societies face a huge challenge in terms of social and medical management of this situation. Although early diagnostic of these diseases has greatly improved in recent years with advances in medical imaging [1] and the availability of biomarkers [2], our ability to prevent, treat or simply slowdown the degenerative processes remains an elusive goal. Intensive research is currently devoted to the deciphering of the cellular and molecular mechanisms underlying neurodegeneration. A more integrated picture has emerged with the recognition of similar and common features between the different neurodegenerative diseases such as the prevalence of protein misfolding and aggregates formation or neuronal death for example. But further advances are crucially needed for a true understanding of the causative factors and the development of efficient treatments.
As an important part of this research, remarkable advances in molecular genetics over the last three decades have allowed many aspects of these diseases to be modelled in animals in order to carry out investigations, which cannot be performed on human beings. A variety of these animal models have been devised with the prominent use of transgenic mice [3], but their repertoire has been largely extended recently to simpler organisms such as the zebrafish Brachydanio rero, the nematode worm Caenorhaditis elegans and the fruit fly Drosophila melanogaster.
This short review, which is not intended to be comprehensive, will be focused on two aspects of this extension. Firstly, I will discuss briefly the key steps in the development of molecular genetics approaches that allowed these new animal models to be devised and studied. I will then examine what has been achieved in this context using D. melanogaster as a model system and the further contributions of this particular organism to the study of human degenerative diseases with the development of new strategies and drugs to tackle these complex pathologies.
A brief overview only of the major results obtained in recent years with these models will be given here and the reader is referred to excellent recent reviews for more detailed accounts on the progress made in modelling specific diseases [4–18].
2 Molecular genetics and the diversification of animal model organisms
The rapid development of molecular biology from the early 1960s led to the emergence of molecular genetics as a new discipline based on a wide range of genetic and molecular tools, which deeply transformed the methodological approaches used in all fields of biological research. In this context, three major technological breakthroughs were instrumental for a great diversification of experimental model organisms.
The first has been the advent of recombinant DNA cloning which made possible to splice DNA fragments of any origin together with a plasmid or viral vector suitable for introduction and amplification in a recipient bacterial host. This gave for the first time an access to the biochemical isolation of genes, their in vitro manipulation or modification and eventually the determination of their complete nucleotide sequence using the DNA sequencing methods, which became soon available. One direct consequence has been a rapid increase in the knowledge of the organization of the genes and the deciphering of the general regulatory mechanisms of their expression with the identification of promoters and tissue-specific enhancer elements. This opened the possibility to devise modular constructs for controlling the expression of cloned genes to be reintroduced in eukaryotic cells.
DNA cloning and engineering technologies paved the way to the development of genetic transformation or transgenesis for the permanent integration of exogenous genetic material into the genome of the germ line of a recipient host and the transmission to its offspring [10,19]. It became thus possible to transfer into the genome of a model organism one of its cloned and engineered genes or a cloned gene from a different organism. This additive transgenic approach was first applied to the mouse in the early 1980s and shortly after to Drosophila [19]. It has remained however limited until recently to a few number of model organisms where its efficiency was high enough for making it practical for the introduction of recombinant constructs with stable germ line transmission.
The development of a site-directed gene editing by homologous recombination in mouse embryonic stem cells gave a very strong impetus to the development of a novel transgenesis approach in the mouse model as it opened the possibility to either inactivate or “knock out” a target gene of interest or replace its coding sequence by “knocking in” a modified copy or the coding sequence of an unrelated gene [20]. This knock out knock in technology, together with additive transgenesis, was used extensively to generate numerous mouse models and a few rat models of Alzheimer's disease, Parkinson's disease, Amyotrophic lateral sclerosis and Huntington's disease which have already provided major insights into the cellular and molecular basis of the degeneration processes (for extensive reviews, see references [3] and [10]).
A third major advance has resulted from the very fast progress in DNA sequencing technologies and bioinformatics which led to the development of the new field of genomics and has given access without any limitation to whole genome sequences of all living organisms. This has been of a great importance regarding animal modelling of human diseases. First, the determination of the sequence of the human genome has allowed the development of genome-wide association studies (GWAS) for uncovering in human populations genetic variations associated with complex diseases thus leading to the identification of novel candidate genes to be examined for their role in the onset and progression of a particular condition [21]. Whole genome sequencing has also revealed the profound degree of evolutionary conservation of the genes throughout all extant organisms of the three domains of the living world, Bacteria, Archea and Eucarya, linked by descent from a common ancestor. Importantly, not only the structure of the encoded proteins is conserved but also in many cases their biochemical function and their interaction with other proteins in molecular complexes or functional pathways recruited in diverse settings during development and differentiation. The most striking example of this evolutionary conservation is that of the Hox genes which play a crucial role in the laying down of the body plan of animal organisms and were first characterized in Drosophila through the identification of the Homeobox, a highly conserved amino acid sequence motif shared by this class of genes [22]. Finally, an important outcome of whole genome sequencing has been to open the possibility to devise short DNA probes to measure accurately and simultaneously the level of expression of all the genes of an organism. Such quantitation can be pushed to the limit of a single cell [23].
3 Advantages and purpose of simpler organisms
Although the mouse has become quickly the dominant model, the new vision of the living world brought about by genome sequencing and the recognition that a major fraction of the genes associated with human diseases are involved in evolutionarily conserved biochemical pathways present in simpler organisms, together with the appreciation of the potency of transgenic gene transfer, has induced a number of researchers to extend the traditional panel of animal model organisms to study human diseases as to include the zebrafish Brachydanio rero, the nematode worm C. elegans and the fruit fly D. melanogaster already used extensively in basic biological research.
Although they cannot be considered as complete alternatives to the mouse, these non-mammalian simpler organisms provide a way to develop complementary strategies for elucidating molecular mechanisms underlying diseases with findings translatable to mammalian systems. Having a short life span and being less expensive and easier to work with, manipulations can be carried out with them much more rapidly than in mice. These organisms have in common to have been extensively used to address general question in developmental and cellular biology and to display versatile panels of powerful genetic tools including transgenesis and genetic screens. The zebrafish has the closest evolutionary relationship to mammals and among its specific advantages is its transparency, which allows live imaging of developmental processes to be performed. Several models of Alzheimer's, Huntington's and Parkinson's disease have been successfully developed in this fish [7,12,18]. The nematode C. elegans offers a number of advantages including a transparent body, a simple and very well defined nervous system and the possibility to carry out extensive forward and reverse genetic screens. It has proved to be a very useful organism for the study of proteinopathy and aging and various models for Alzheimer's disease, Huntington's disease, Parkinson's disease, Amyotrophic lateral sclerosis, taupathies and fronto temporal dementia have been modeled in this organism [4,16,24,25].
Among these three animal models, D. melanogaster stands out as an invaluable organism to study human diseases [26–28].
4 Drosophila melanogaster as a model organism of choice to study neurodegenerative diseases
Drosophila has been used as a laboratory animal for over one hundred years and has made a number of major contributions to the understanding of fundamental biological principles. This includes the nature of genetic mutations, the structure of chromosomes, the demonstration of the chromosomal theory of inheritance and the functional definition of genes. In a more recent period it has contributed to the identification of genes involved in a number of biological processes such as embryonic development and patterning, innate immunity, the ontogeny of the nervous system for example. Its development is well characterized and the mechanisms and pathways involved have been largely conserved during evolution. It has a high biological complexity, for example its nervous system is composed of approximately 200,000 neurons as compared to only 302 in C. elegans. It displays a complex behavior and memory.
Drosophila has a number of practical advantages as an experimental organism such as the ease and inexpensive cost of breeding, a short life cycle and large progeny. Most importantly, sophisticated genetics tools are available which are constantly being improved and disseminated in the scientific community [26]. This includes transgenesis and powerful genetic systems for manipulating gene expression such as the UAS-GAL4 system which offer the possibility to target and control in time and space the expression of transgenes in specific tissues including constructs coding for toxins for cell ablation [29]. Another important tool is a system for the generation of mosaic tissues in which clones of cells, which can be as small as a single neuron, are made homozygous for a mutation in an otherwise heterozygous background [28]. Most prominent is the availability of a wealth of materials and resources such as freely available large collections of Drosophila stocks carrying transgenic constructs or loss or gain of function mutations affecting a large fraction of the genes. This makes Drosophila a very versatile model organism to study genetic, molecular and cellular processes in most fields of biology such as development, morphogenesis, cell polarity, memory, behaviourly, endocrine control of metabolism, innate immunity, neurogenesis, biology of stem cells, differentiation of tissues and organs.
In addition to these advantages, the use of Drosophila as an experimental organism does not raise ethical problems comparable to those of experimental manipulations on rodents or other mammalian models including non-human primates.
The sequencing of the Drosophila genome in 2000 has revealed that at least 77% of the human disease-associated genes reported in the Online Mendelian Inheritance in man database have evolutionary similarities with Drosophila genes [26,30]. Importantly, these genes include most of the genes known to be associated with familial forms of neurodegenerative diseases.
5 Drosophila and the study of neurodegeneration
Historically, the interest for using D. melanogaster to study neurodegeneration can be traced to the earlier studies carried out by Benzer and his colleagues who pioneered behaviourly genetics by the isolation of phototactic mutants. This opened the way to the identification of Drosophila genes involved in brain function and integrity, aging and degeneration [31]. Subsequently, the first transgenic fly models of neurodegenerative diseases were devised in 1998 with a model of spinocerebellar ataxia 3 (SCA3) [32] and a model for Hunttington's disease [33] based in both cases on the expression of a protein with an expanded polyglutamine repeat, which is considered as the primary pathogenic factor in the onset of these diseases.
The creation of new and more sophisticated models has been a continuous trend since then by taking advantage of the powerful resources of Drosophila genetics, which allow four complementary approaches to be applied individually or in a combined manner.
The first is the isolation of loss or gain of function mutations in forward genetic screens aiming at the isolation of mutants with impaired nervous system maintenance and functional integrity [34]. These mutations can be generated by chemical mutagens or transposon mobilization, which provides means to disrupt or increase the expression of specific genes. More recently gene expression knock down by short double stranded RNA interference (RNAi) has brought about the possibility to carry out large-scale functional screens by conditional disruption of gene expression [28,35]. In addition, the advent of the CRISP/Cas9 technology based on the use of bacterial RNA-guided nucleases has provided an invaluable tool for the controlled and targeted edition of any gene with an efficiency and precision never attained before [36]. A variety of phenotypes can be used for these screens, which include behavioral defects, shortened lifespan, CNS degeneration analyzed by histology or electrophysiology, paralysis and altered locomotion or disturbed circadian rhythm.
An alternative “reverse genetics” or candidate gene approach, which has been extensively used, is to isolate loss of function mutation in or induce RNAi knocked down expression of the fly homolog of a human gene presumed to be implicated in a neurodegenerative disease [37] and evaluate the phenotypic consequences.
The third approach makes use of the ease of transgenesis in Drosophila which prompted a number of research groups, after the initial modeling of the polyglutamine repeat SCA3 and HD diseases, to use the fly to assess the effect of the expression of wild type or mutated versions of human genes involved in neurodegenerative diseases integrated permanently in the Drosophila genome. These studies relied heavily on the use of the UAS-Gal4 system to direct the expression of the transgenes in neuronal tissues of the fly [29]. One organ of choice for such a targeted expression is the compound eye. The Drosophila visual system has been the subject of numerous studies pioneered by Seymour Benzer [31]. The structure of the fly eye makes it ideally suited as a read-out for detecting any changes induced by the expression of transgenic foreign genes. It is composed of approximately 750 individual visual receptors called ommatidia each made up of eight photoreceptor cells and 11 accessory cells. The ommatidia are arranged in a highly regular repetitive array and any perturbation in the pathways of ommatidia development or affecting the shape, size and arrangements of the cells induce alterations of this regular structure that are easily detectable as a typical rough aspect of the surface of the eye. This approach is illustrated by the targeted expression in the fly eye of the human Aβ amyloid peptide, which accumulates in neurons of Alzheimer patients (Fig. 1). The severity of the rough eye phenotype is correlated with the level of expression and provides also an indication of the toxicity of the expressed proteins.
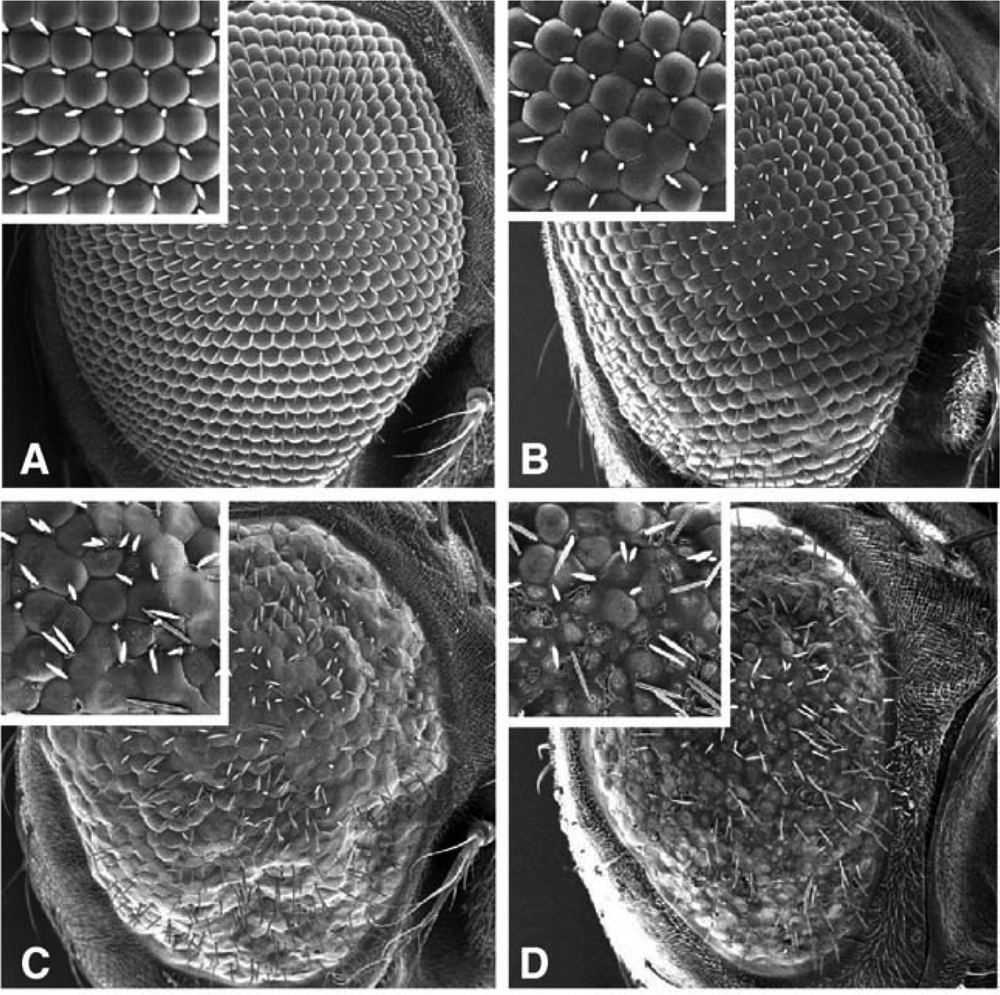
Rough phenotype induced in the Drosophila melanogaster eye by the expression of the amyloid beta (Aβ) peptide causing neurodegeneration in Alzheimer patients. A: wild type eye. B, C, D: increased severity of the rough eye phenotype paralleling the increase of expression of the Aβ transgene.
Reprinted from A. Finelli, A. Kelkar, H.J. Song, H. Yang, M. Konsolaki, A model for studying Alzheimer's Aβ42-induced toxicity in Drosophila melanogaster, Mol. Cell. Neurosci. 26(3) (2004) 365–375, with permission from Elsevier.
Besides eye roughening, a large panel of other phenotypic traits can be analyzed such as neurotoxicity, locomotion defects, alteration of synaptic transmission, reduced life span, intracellular and extracellular accumulation of proteins and aggregates formation, fibrillar tangles deposits, memory tests, circadian rhythm.
The number of transgenic models of overexpression of disease-related proteins has been greatly expanded as to include a large panel of diseases and proteinopathies [5,8,10] including Alzheimer's disease [15], Parkinson's disease [38], polyglutamine expansion disorders such as Hunttington's disease [14], spinocerebellar ataxias [11], amyotrophic lateral sclerosis [6].
The fourth approach exploits one of the greatest strengths of Drosophila, which is the possibility to carry out genetic screens for uncovering genetic interactions taking place between two genes. In this approach, second sites genetic modifiers are selected to identify the genes whose mutation or knocked down expression enhances or suppresses a given phenotype induced by a first mutation. The interest of such suppressor–enhancer screens is that they can be performed in an unbiased manner without any preconception on the biological function of the modifiers to be found. They can be applied to identify modifiers of various relevant phenotypes such as the neurotoxicity of a transgenic construct as described in Fig. 2 [15,26].
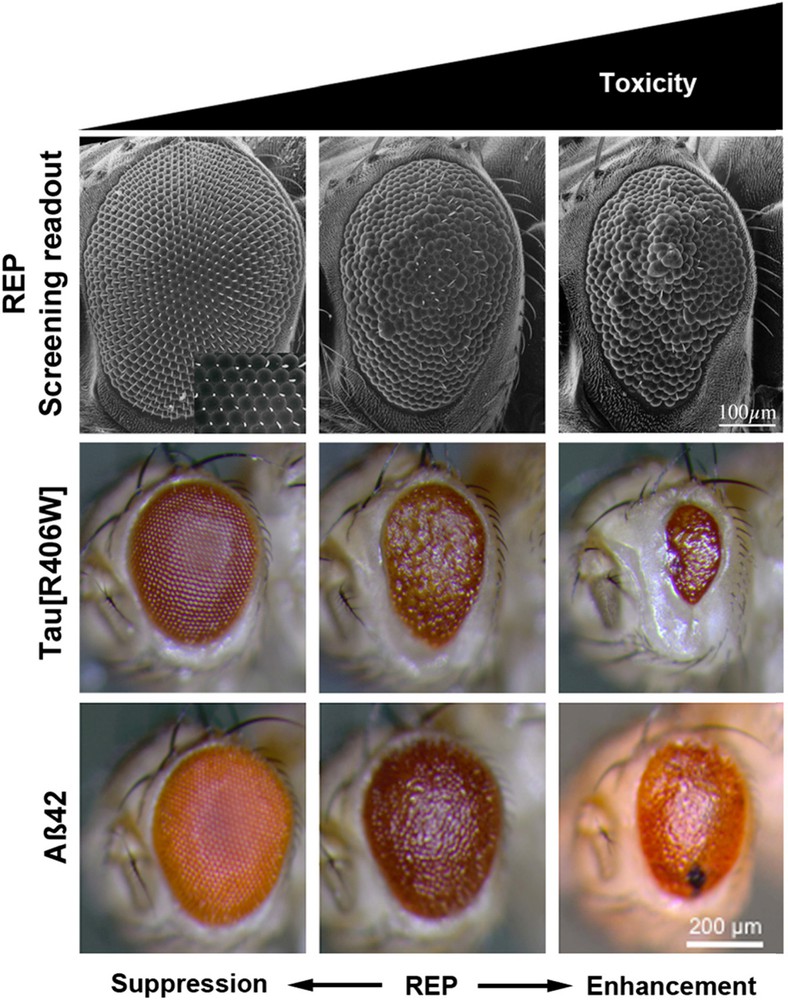
(Color online.) Examples of rough eye phenotype (REP) used for screening enhancers (right panel) or suppressors (left panel) of neurodegeneration induced by expression of transgenes coding a variant of the human tau protein or the amyloid Aβ peptide.
Reprinted from K. Prüßing et al., Drosophila melanogaster as a model organism for Alzheimer's disease, Mol. Neurodegener. 8 (2013) 35–46.
6 What has been found using Drosophila?
The application of the various strategies described above, often used in combination, has led to the generation of a large number of fly models for the study of neurodegenerative diseases.
Firstly, the forward and reverse genetic screens have brought an important contribution to the identification of Drosophila genes involved in the integrity and maintenance of the nervous system. Earlier forward genetic screens for recessive loss of function mutations in fly genes have helped discover more than 40 genes, which are orthologues of human genes [34]. More than half of these fly genes are related to human genes, which have been associated with neurodegeneration. They are involved in a variety of basic cellular processes and functions such as signaling pathways, mitochondrial, microtubules and actin skeleton functions, the control of lipid homeostasis and protein homeostasis by the ubiquitin-proteasome degradation machinery or autophagy and lysosomal degradation [31,34]. This list underestimates the actual number of genes whose mutation may lead to a loss of integrity or function of the fly nervous system. These screens were not devised primarily for the isolation of mutations causing neurodegeneration and they may have thus missed a much larger number of genes whose mutation impairs neuronal functions but does not result in a conspicuous or easily scorable phenotype at the level of the whole fly. This is confirmed by a recent mosaic screen of the Drosophila X chromosome aiming at identifying novel genes required for the development, function and maintenance of the nervous system [28].
The reverse or candidate gene approach has provided an important contribution to the understanding of Parkinson's disease pathogenesis through the demonstration that mutations of the fly homologues of the parkin and pink1 genes which had been associated with familial forms of PD, disrupt mitochondrial integrity and function [38].
Screens for modifiers of neurodegeneration or neurotoxicity phenotypes induced by a transgenic construct expressing a wild type or a mutated human protein have proven to be very powerful for the identification of genes contributing either globally or more specifically to the susceptibility to neurodegenation. Two examples underline the interest of this approach for pointing out to genetic interactions that in some cases were not suspected to take place in the degeneration process from previous human genetic studies.
In a functional screen for the identification of Alzheimer's disease susceptibility genes, RNAi-mediated loss of function mutations in 87 fly genes homologous to human genes associated with Alzheimer’ disease in genome-wide association studies were tested for the modulation of neurotoxicity induced by the expression of the human Tau protein which may mediate the deleterious effects of the accumulation of the amyloid-β peptide in neurons of AD patients [37]. This led to the identification of several modifiers genes, which reveal the implication of cell adhesion pathways in Tau toxicity [37].
A genome-wide screen for modifiers of degeneration induced by the expression in the fly eye of a transgenic construct containing an expanded polyglutamine domain in the ataxin-3 gene as a model of type-3 spinocerebellar ataxia, led to the identification of a series of modifiers belonging to the chaperone and ubiquitin-proteasome pathways. Interestingly, in addition to these modifiers of protein folding and degradation, other modifiers isolated in the same screen modulated also Tau toxicity, revealing that there are common pathways in neurodegeneration due to distinct human neurotoxic proteins [39].
Transgenic models have diversified and improved so as to reproduce some of the features of the onset and progression of the disease in human patients. A fine example is provided by the recent development of an aged onset of AD. Earlier transgenic studies had confirmed the “amyloid” hypothesis that the accumulation and aggregation in neurons of the Aβ42 peptide generated by proteolytic cleavage of the Amyloid precursor protein (APP) is the primary cause of neurotoxicity and is upstream of a cascade of molecular and cellular events leading to AD and neuronal death [40]. For this new model, a transgenic fly stock was established harboring two transgenes whose expression is driven specifically in the central nervous system [41]. One of the transgenes expresses a low level of human APP while the other expresses the beta-site APP cleaving enzyme BACE whose substrate is APP. Remarkably, adult flies show an age-dependent decrease in motor reflex behavior, an increase of Aβ peptide load, neuroanotomical defects, memory deficit and synaptic dysfunction [41]. Expression of APP and BACE in larvae leads to defects in neuromuscular junction, defective locomotion behavior and several synaptic abnormalities [42].
Another example of the power of the transgenic approach is provided by a recent study of the intrinsic toxicity of naturally occurring fragments of the Hunttingtin protein. These peptides have been implicated by clinical studies and animal models in the pathogenesis of the Huttington's disease, which results from the expansion of a polyglutamine domain located in the N-terminal region of the protein. The pathogenic effects of the expression of seven Hunttingtin fragments expressed in different Drosophila transgenic lines were compared using the criteria of survival to adulthood, degeneration of neurons, motor function and longevity. The results of these in vivo tests combined with a study of the biophysical properties of the peptides converge to the conclusion that the short peptide coded by exon 1 of the Hunttingtin gene is exceptionally pathogenic [43], making it a new target for a potential therapy.
7 Future prospects for the fly model
The various approaches described above can be integrated in a conceptual framework (Fig. 3) whose cogency for an application to all human neurodegeneration diseases is supported by the wealth of contributions accumulated with the transgenic fly models. It can also be applied to a wider range of human diseases as shown for example by advances in modeling myotonic dystrophy [44], Friedreich's ataxia [45], spinal muscular atrophy (SMA) [46], oculo pharyngal muscular dystrophy [47], amyotrophic lateral sclerosis [48], and prion disease [5].
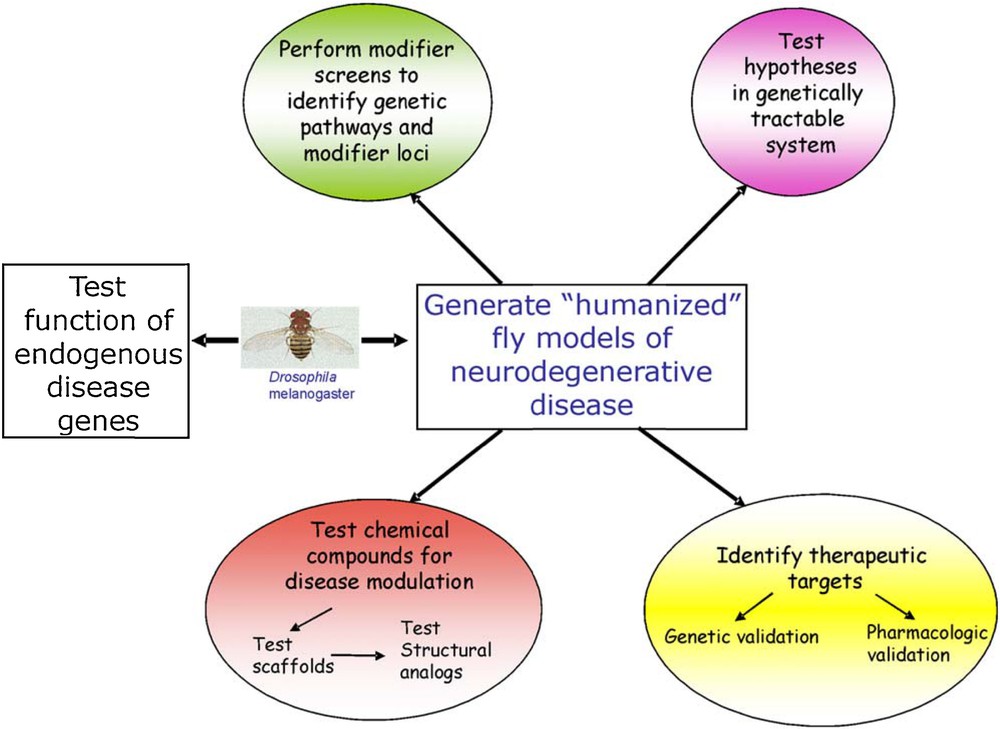
(Color online.) Strategies for using Drosophila melanogaster as a model organism for the study of human neurodegenerative diseases.
Reprinted from J.L. Marsh and L.M. Thomson, Drosophila in the study of neurodegenerative disease, Neuron 52 (2006) 169–178, with permission from Elsevier.
Some limitations of the fly as a biological organism for modeling human diseases should however be recognized. They are similar to that of C. elegans and zebrafish. Although basic neuronal processes involve evolutionary conserved molecules and signaling or developmental pathways, the human nervous system has a much higher degree of organizational and functional complexity than that of these simpler organisms. It is not thus surprising that none of the “humanized” fly models recapitulates the full range of features of the human neurodegenerative diseases. Because of the large evolutionary distance between arthropods and vertebrates, it is also possible that some biochemical functions of the degeneration-related proteins under study have largely diverged, rendering vertebrate-specific a number of disease causative factors and thus impossible to identify and reproduce in the fly model. Results obtained with the various Drosophila models should thus be translated or validated in mammalian systems such as mice, other rodents and non-human primates before they are validated eventually in human patients.
Despite these limitations, Drosophila stands out as a powerful organism to address directly a number of questions pertaining to the mechanisms and progression of neurodegeneration. Specific hypotheses can be tested and novel therapeutic targets revealed, which can be further validated in mice and other genetically tractable organisms such as C. elegans and zebrafish.
The prominent strength of the fly resides in the fast advance in the functional knowledge of its genes and biochemical pathways which progresses steadily towards a degree of completion not yet reached by any other organism. This trend is powered in particular by the efficiency of the genetic tools, which allow highly specific and sophisticated genetic modifier screens to be devised. Besides the identification of novel modifier genes, unbiased genetic screens for cost-effective drug discovery can be performed for testing the protective role of a variety of compounds including small molecules, which could be tested for therapeutic use in further appropriate models [49–51]. This is illustrated by three recent examples. One is the finding that lithium suppresses the pathological effects of accumulation of the Aβ peptide in a model of Alzheimer's disease by reducing protein translation [52]. Another is the demonstration of the therapeutic potential of methylene blue that has been shown to reduced neurodegeneration and Hunttingtin aggregation in a fly HD model [53] and rescue heart defects in a fly model of Friedreich's ataxia [45]. The third one is the demonstration of the protective role against the aggregation of the polyglutamine disease protein of a short peptide from Huttingtin in an eye-toxicity test [54].
Considering that D. melanogaster probably stands presently as the best-analyzed and understood multicellular organism to date, it can be envisaged that it will stay for the years to come at the forefront of the most extensively used organisms for unravelling the molecular mechanisms underlying the devastating neurodegeneration diseases.
Disclosure of interest
The author declares that he has no conflicts of interest concerning this article.