

1 Introduction
Les rétinopathies pigmentaires, qui affectent 30 000 à 40 000 personnes en France, figurent au sein d'une large liste d'affections orphelines, au sein desquelles elles se singularisent par une hétérogénéité tant génotypique que phénotypique extrême.
Voilà à peine une quinzaine d'années, une liste très courte de loci impliqués dans la transmission de ces maladies (autosomale dominante ou récessive ou liées au chromosome X) n'attirait l'attention que de quelques rares généticiens, et encore plus rares ophtalmologistes. Une consultation typique pour un patient atteint de rétinopathie pigmentaire se déroulait de la façon suivante : le patient, souvent un jeune adulte, avait ressenti dès l'enfance des troubles d'adaptation à l'obscurité longtemps méconnus, puis un rétrécissement du champ visuel, conduisant parfois à un incident révélant la maladie ; cela aboutissait progressivement, dans un délai de 15 à 20 ans, à la perte de la vision centrale et donc à la cécité (Fig. 1). Aucune perspective de stabilisation, et encore moins de guérison, ne pouvait être évoquée avec ces familles.
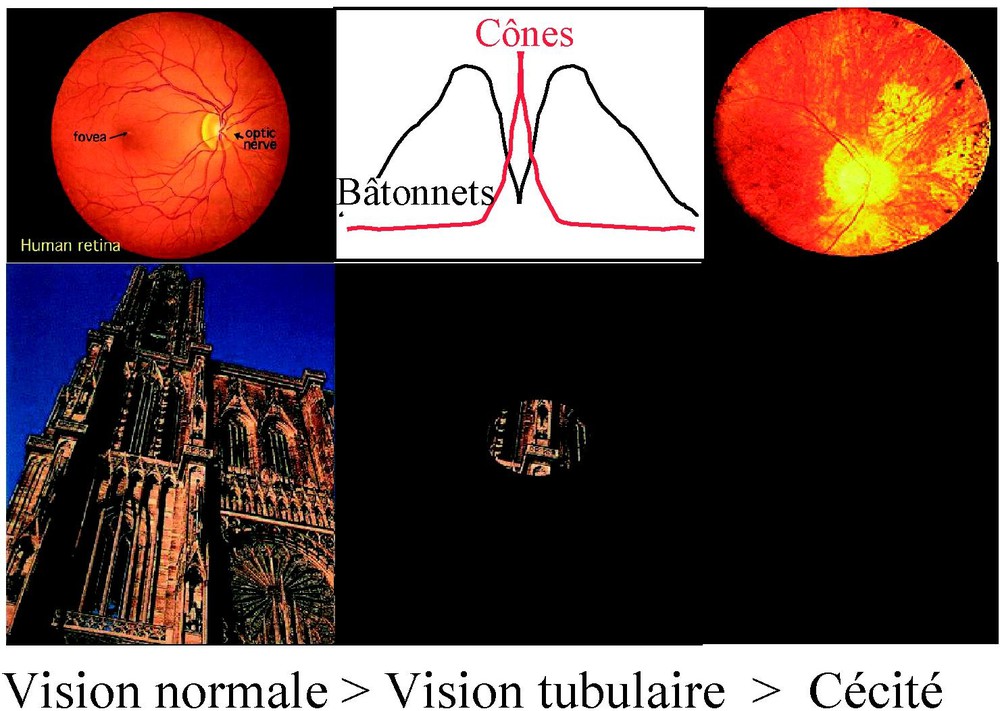
Perte de la vision centrale conduisant à la cécité.
La découverte d'un nombre croissant de mutations des gènes appartenant à de multiples familles a aujourd'hui pour conséquences, d'une part, l'absence de diagnostic génétique systématique et, d'autre part, comme implication la nécessité tout au moins économique de concevoir des approches thérapeutiques communes à plusieurs types de mutations.
2 Progrès en physiopathologie
La rétine comporte deux systèmes fonctionnant dans des conditions différentes. En lumière scotopique, donc dans la pénombre, les photorécepteurs à bâtonnets permettent de détecter des intensités lumineuses très faibles. Ce système ne possède pas une capacité de résolution importante et ne contribue pas à l'acuité visuelle. Il est essentiel à la vision périphérique et à la vision nocturne. Le système des photorécepteurs à cônes et la circuiterie rétinienne qui lui correspond prédominent en rétine centrale, au niveau de la macula. Il assure non seulement la vision colorée mais aussi la vision à haut contraste, l'acuité visuelle et toutes les fonctions visuelles en atmosphère lumineuse normale. Ce dernier système, dans les conditions actuelles d'éclairement, est largement prédominant.
À partir du début des années 1990, la découverte de mutations à l'origine de la rétinopathie pigmentaire a progressé à une vitesse impressionnante. Aujourd'hui, plus de 140 loci sont impliqués dans la genèse de ce groupe de maladies, dont seulement la moitié a été caractérisée. Le premier gène identifié a été celui de la rhodopsine, proteine initiant la transduction visuelle au niveau du segment externe des bâtonnets. Environ 90 % des mutations identifiées s'expriment principalement dans les bâtonnets au niveau de la transduction ou de protéines de structure. Ce défaut génétique explique parfaitement la perte de la vision périphérique et surtout nocturne des patients. Le mécanisme de dégénérescence des bâtonnets par apoptose a été démontré en 1993. Le lien entre les mutations et l'apoptose n'est pas élucidé. Quant à la mort des cônes, à l'origine de la perte de vision centrale, aucune hypothèse et aucun mécanisme n'avaient été jusqu'ici validés. Rien ne permettait d'expliquer la perte secondaire de fonction des cônes, responsable de la disparition tardive de la vision centrale, donc de la cécité.
3 Recherches thérapeutiques
3.1 Thérapie génique
La conséquence la plus directe de la découverte de mutations causales a été d'ouvrir des perspectives en thérapie génique. De telles approches ont été entreprises depuis maintenant plusieurs années dans certains modèles animaux, correspondant de manière assez proche à la maladie humaine, avec une restauration anatomique et fonctionnelle partielle. Tout récemment, un travail marquant a été réalisé sur des chiens porteurs d'une maladie proche de l'amaurose congénitale de Leber, la plus grave des formes de rétinopathie pigmentaire, puisqu'elle conduit à la cécité dès la naissance. La réintroduction d'un gène exprimé, cette fois-ci, dans l'épithélium pigmentaire, et impliqué dans le métabolisme de la vitamine A, et donc des pigments visuels, a permis chez trois chiens de restaurer des fonctions électrophysiologiques partielles, mais significatives, et une compétence visuelle utilisable par l'animal [1]. La restauration fonctionnelle est donc possible par thérapie génique. Restent cependant de nombreux écueils : établissement de sa sécurité, multiplicité des mutations et donc des approches à envisager, sans parler des problèmes posés par les mutations dominantes.
3.2 Neuroprotection de l'ensemble des photorécepteurs
À côté de la thérapie génique de correction du défaut génétique, plusieurs stratégies ont été conçues pour limiter ou bloquer la perte des photorécepteurs. Leur ensemble peut être englobé sous le terme de neuroprotection. Elles font appel à la pharmacologie [2], à l'usage de facteurs de survie cellulaire, aux approches anti-apoptotiques et à la thérapie cellulaire.
La démonstration du rôle neuroprotecteur d'un facteur de croissance, le fibroblast growth factor (FGF), dans un modèle animal de rétinopathie pigmentaire, le rat RCS, remonte déjà à plus d'une dizaine d'années [3]. Nous avons démontré le bénéfice structural et fonctionnel apporté par des injections sous-rétiniennes de GDNF (Glial-cell line derived neurotrophic factor) [4]. Un autre facteur, le ciliary neurotrophic factor (CNTF) a aussi été utilisé avec succès dans plusieurs modèles animaux, et des essais cliniques sont en cours, faisant appel à la délivrance par des cellules encapsulées, libérant le facteur neurotrophique [5].
3.3 Neuroprotection des photorécepteurs à cônes
Le fait que dans le tableau clinique humain, et dans la plupart des modèles animaux, la disparition des cônes survienne après celle des bâtonnets, nous a conduits à envisager plusieurs hypothèses, parmi lesquelles nous en avons validé deux : celle d'une neurotoxicité initiale lors de la dégénérescence des bâtonnets, et surtout celle d'une dépendance trophique des cônes sur les bâtonnets. La transplantation de bâtonnets préparés en couche pure au vibratome, dans un modèle animal porteur d'une rétinopathie pigmentaire très brutale proche de certaines formes humaines, a été réalisée à un âge où l'ensemble des bâtonnets a dégénéré, et où les cônes ont seulement entrepris leur processus de dégénérescence. L'intervention ralentit de moitié la dégénérescence des cônes [6].
Sur des modèles de cocultures, cet effet de survie exercé par le transplant de bâtonnets sur les cônes s'avère lié à la libération de molécules diffusibles de nature protéique. Il est important de souligner que cet effet s'exerce aussi longtemps que les bâtonnets, y compris ceux affectés par une mutation, sont viables [7,8].
Il est donc vraisemblable que la disparition secondaire des cônes soit un événement lié à la privation d'un facteur de viabilité cellulaire. Cette constatation nous a conduits :
- − à envisager un protocole de recherche clinique sur la transplantation de photorécepteurs chez des patients ayant atteint le stade très tardif d'une rétinopathie pigmentaire ;
- − à caractériser la ou les molécules impliquées dans la signalisation qui assure la viabilité des cônes.
Pour identifier les RdCVF, nous avons développé une approche systématique par clonage par expression basée sur un test de viabilité des cônes (Fig. 2). Le modèle utilisé est une culture de cellules rétiniennes d'embryons de poulet, cultivées à faible densité et en l'absence d'agents inducteurs, ce qui conduit à une différentiation en photorécepteurs qui sont majoritairement des cônes, contrairement à ce qui est observé dans des cultures de cellules de mammifères pour lesquels les cônes ne représentent que 3 à 5 % des photorécepteurs. La dégénérescence des neurones rétiniens aviaires, cellules post-mitotiques survient sur une période de plusieurs jours. La dégénérescence est ralentie lorsque ces cultures sont réalisées en présence de milieu conditionné préparé à partir d'explants rétiniens de souris normales. Ceci démontre qu'une molécule synthétisée et sécrétée par les rétines de souris est capable d'induire une survie des cônes de poulet. Une banque d'expression de rétine de souris a été construite. Les plasmides de cette banque ont été transfectés dans des cellules COS, d'abord par ensemble de 100 clones, et les milieux conditionnés issus de ces cellules transfectées ont ensuite été récoltés et transférés dans les puits des cultures de cônes. Après incubation pour une période de sept jours, la viabilité a été mesurée par comptage cellulaire à l'aide de sondes fluorescentes et par utilisation d'un microscope piloté par un logiciel adapté. Parmi 2100 ensembles testés, un ensemble de 100 clones, qui présentait la plus grande amplitude de l'effet neuroprotecteur, a été divisé en sous ensembles de 10 clones, qui ont ensuite été testés. Cette procédure de dilution limitée a conduit à l'isolement d'un clone présentant une phase de lecture correspondant à une nouvelle protéine de 109 acides aminés, que nous avons nommé rod-derived cone viability factor [9].
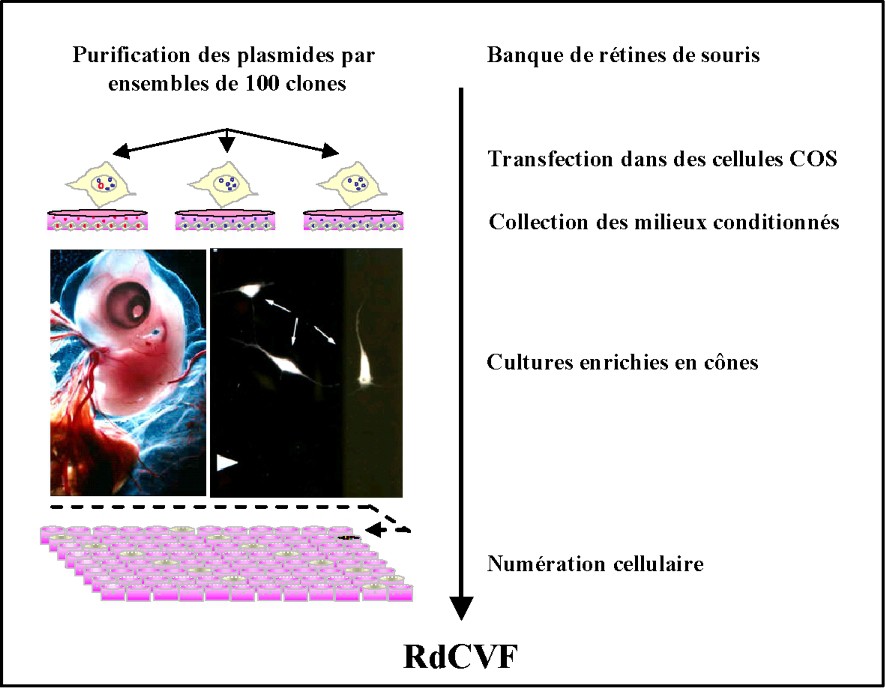
Approche systématique par clonage par expression basée sur un test de viabilité des cônes permettant d'identifier les RdCVF.
RdCVF présente une homologie avec la famille des thioredoxines. Le polypeptide isolé est un variant d'épissage d'un nouveau gène (Txnl6), troqué dans le motif thiorédoxine et ne possédant pas l'activité thiol-oxydoréductase. Il existe pourtant un second messager présentant un motif thiorédoxine entier et donc probablement l'activité enzymatique (Fig. 3). Txnl6 est donc potentiellement un gène bi fonctionnel avec une forme correspondant au facteur de viabilité des cônes et l'autre à une thiol-oxydoréductase. L'homologie avec la famille des thioredoxines est intéressante, car bien que RdCVF ne possède pas d'activité enzymatique, les thioredoxines sont une famille de protéines sécrétées présentes chez tous les organismes et ayant des propriétés pleiotropiques, dont la régulation du potentiel redox de la cellule n'est qu'une des multiples fonctions. Le prototype de la famille, Trx1, a d'ailleurs les propriétés d'une cytokine. RdCVF présente de plus une analogie avec Trx80, une forme tronquée de Trx1, qui n'a pas non plus d'activité enzymatique et qui présente une activité cytokine distincte de Trx1 [10]. L'expression des deux messagers de RdCVF, qui est spécifique à la rétine, est perdue après la perte des bâtonnets. La protéine est retrouvée dans les segments externes des bâtonnets et dans l'espace extracellulaire entourant les photorécepteurs (Fig. 4). À plus haute résolution, un marquage plus intense, colocalisant avec la surface des cônes suggère l'existence d'un site d'affinité pour RdCVF à la surface des cônes. Au plan fonctionnel, l'injection de RdCVF purifié dans l'espace sous rétinien de souris rd1 après dégénérescence des bâtonnets aboutit à une protection significative des cônes sur une période d'une quinzaine de jours. In vitro, l'élimination par immunodéplétion de RdCVF résulte en une forte diminution de l'effet protecteur induit par le milieu conditionné ce qui implique que la signalisation bâtonnets-cônes, au moins dans ces conditions expérimentales, dépend de RdCVF.
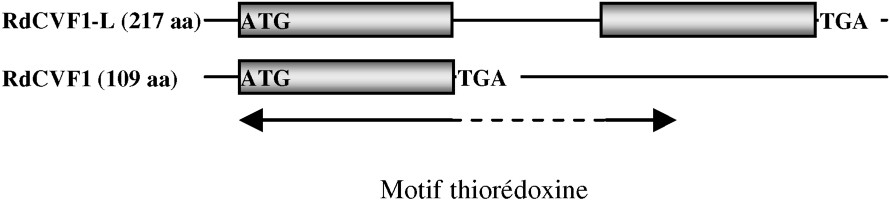
Messager présentant un motif thiorédoxine entier et donc probablement l'activité enzymatique.
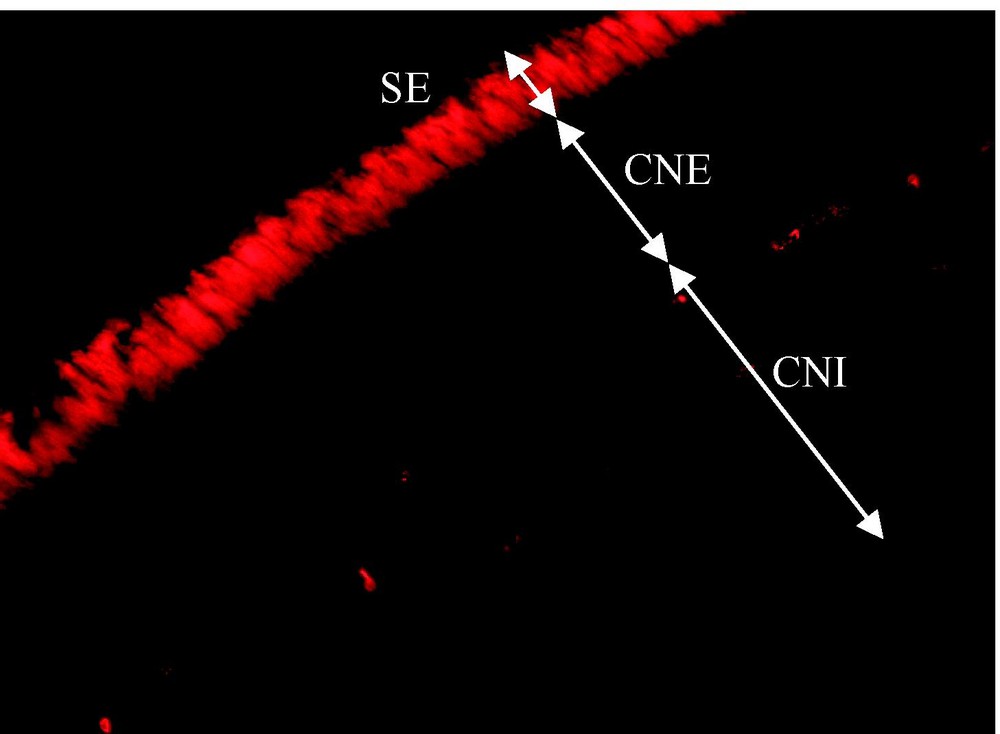
Segments externes des bâtonnets et espace extracellulaire entourant les photorécepteurs.
L'approche systématique de criblage des facteurs de viabilité des cônes utilisant un modèle cellulaire a conduit à l'identification d'une protéine non précédemment répertoriée dans les facteurs de croissance ce qui constitue a posteriori une validation de notre approche expérimentale privilégiant la fonction. Les mécanismes par lesquels RdCVF exerce un effet protecteur sur les cônes, comme ceux sous-tendant la signalisation par Trx80, sont inconnus. Ayant identifié un acteur de l'interaction entre les bâtonnets et les cônes, l'étude de la signalisation sous-jacente devient envisageable. L'inactivation du gène par recombinaison homologue et l'utilisation de l'interférence ARN sont mises en oeuvre pour confirmer le rôle essentiel de RdCVF in vivo.
RdCVF, un facteur sécrété par les bâtonnets nécessaire à la survie des cônes et représente une nouvelle piste thérapeutique pour les rétinites pigmentaires. La question de son mode de délivrance est au cœur de nos projets actuels, qu'il s'agisse d'administration par le moyen de vecteurs viraux ou de cellules encapsulées. Ces outils permettront, dans une première phase du transfert vers la clinique, de tester le bénéfice sur la fonction visuelle à long terme chez des modèles animaux de rétinopathies pigmentaires tels que les rongeurs, mais aussi chez des animaux plus proches de l'homme, tel le chien ou le porc. Ces étapes sont nécessaires avant d'envisager un essai clinique pour lequel la population devra être soigneusement sélectionnée suivant des critères cliniques et génétiques. Toutes ces considérations, ne permettant une éventuelle application chez l'homme que dans quelques années, sont à mettre en perspective de l'espoir suscité par la déclaration d'Alan Wright : Preserving cones would prevent 1.5 Million people worldwide from becoming blind [11].
4 Conclusion
Le décryptage des anomalies génétiques sous-jacentes au développement des rétinopathies pigmentaires a permis de concevoir des hypothèses physiopathologiques, d'utiliser des modèles existants, ou d'en créer, d'identifier des voies conduisant à la mort cellulaire, et de proposer des approches permettant de limiter cette perte cellulaire, tant au niveau des bâtonnets que des cônes. La préservation de la fonction des cônes représente un enjeu majeur, dont la portée englobe vraisemblablement la principale cause de cécité de l'adulte : la dégénérescence maculaire liée à l'âge. Une population de 10 % des cônes pourrait suffire à mener une vie normale dans nos environnements lumineux. Il ne s'agit certes pas d'une approche curative de la maladie, mais dans une affection où aucune perspective thérapeutique n'a jamais pu être proposée, cette préservation représente déjà potentiellement un progrès. Les approches ne seront pas uniformes et l'identification des mutations spécifiques en sera l'élément dominant. À ce jour, en effet, l'évolution naturelle des rétinopathies pigmentaires n'a pas encore été améliorée par les progrès conceptuels que nous avons résumés. La perspective de prolonger l'usage d'une vision centrale permet cependant, aujourd'hui, de tenir au patient un discours crédible d'encouragement à la réadaptation sensorielle utilisant la vision résiduelle, en attendant de pouvoir mettre en œuvre les essais thérapeutiques visant à la préserver.
Acknowledgments
David Hicks, Olivier Poch, Serge Picaud et Georges Lambrou. Ces travaux de recherche bénéficient du soutien de l'Inserm, l'AFM (Association française contre les myopathies), de l'association Retina France Vaincre les maladies de la vue, de la Fédération des aveugles et handicapés visuels de France, de la FRM, fondation pour la recherche médicale, du prix coup d'Elan de la fondation Bettencourt-Schueller, de la fondation américaine Foundation Fighting Blindness et de Novartis Pharma.