

1 Introduction
Nucleocytoplasmic transport is a vital process for nucleated cells, which need to regulate the flow of macromolecules transiting between the cytoplasm and the nucleus [1–4]. Molecules larger than 50 kDa are usually transported across the nuclear envelope via an active process able to pump macromolecules against a concentration gradient. Both nuclear import and export take place through the large nuclear pore complexes spanning the nuclear membrane. However, a different set of soluble receptors is required in each case. In animal cells, the main import pathway involves the two soluble receptors importin α and importin β. These bind to import cargoes carrying short amino acid sequences known as nuclear localization signals (NLSs), forming a heterotrimer called the cargo complex. This complex is subsequently able to find its way to the nuclear envelope and through a nuclear pore complex. The translocation is facilitated by the interaction of importin β with protein components of the nuclear pore containing phenylalanine–glycine repeats.
Once in the nucleus, the cargo complex is dissociated by the GTP-binding protein Ran, which in its nuclear Ran-GTP form binds importin β with high affinity [5]. After cargo release, the Ran/importin β complex is transported back to the cytoplasm, where the cytoplasmic protein RanBP1 catalyzes the hydrolysis of RanGTP into RanGDP. This causes Ran to change conformation, leading to the release of importin β in the cytoplasm. Importin α is also actively re-exported back to the cytoplasm, after forming a complex with the soluble export factor CAS and RanGTP. The transported cargo, on the other hand, remains in the nucleus, free to carry out its nuclear function but unable to traverse the nuclear pore in the other direction. The cycle is completed with RanGDP being returned to the nucleus via association with the soluble import factor NTF2, and there is an undergoing nucleotide exchange for GTP supported by the chromatin associated protein RCC1. This permits the restoration of the nuclear pool of RanGTP. Indeed, the different interactions of Ran with the numerous Ran-binding proteins present in the cytoplasm and in the nucleus result in a primarily nuclear localization of the protein during interphase, as well as in an asymmetric distribution of the two different forms of Ran: the predominant form of Ran in the nucleus is RanGTP, while the predominant form of Ran in the cytoplasm is RanGDP [6]. This RanGTP to RanGDP gradient across the nuclear membrane thus ultimately drives vectorial nuclear transport.
Interestingly, the same set of soluble factors that are involved in nuclear import have been shown to intervene in a number of other different cellular processes [7]. During mitosis, the importin α/β complex inhibits spindle assembly factors by binding to them, while RanGTP, by releasing these factors from the α/β complex, promotes spindle assembly [8,9]. It has also been proposed that RanGTP, which is expected to remain associated with chromatin during interphase, may control other chromatin related processes, for example, the assembly of the nuclear membrane around the chromatin as well as assembly of the nuclear pore complexes [10,11]. In addition, recent evidence links Ran and importin β to the control of the duplication of the microtubule organizing center [12]. Finally, RanGTP and importin β seem to have a connection to gene transcription, since the former is found associated with transcriptionally inactive and the latter with transcriptionally active genes on the chromatin [13]. A common theme in many of these cellular processes could be the capacity of RanGTP to act as a molecular switch by turning on or off the interactions between the importin α/β heterodimer and different key targets.
This molecular switch capacity is enabled by the clever pattern of interactions existing between nuclear localization signals, importin α, importin β and Ran. These interactions (summarized in Fig. 1) have been understood through a series of structural and biochemical studies [14–18]. The recognition of a NLS by importin α occurs only in presence of importin β. This is because importin α itself carries at its N terminus an auto-inhibitory NLS-like domain rich in basic amino acids, which competes with the binding of other NLSs to the protein (Fig. 1a) [16]. This auto-inhibitory domain is also the importin β binding (IBB) motif of importin α. As a result, when importin β binds importin α, it sequesters the auto-inhibitory domain, exposing the NLS binding groove and allowing NLS-carrying proteins to interact with importin α (Fig. 1b). On the other hand, the binding of RanGTP to importin β competes with the binding of importin β with importin α, since on importin β the binding domains for importin α and RanGTP partially overlap. So the effect of RanGTP is to release the cargo from the importin α/β complex. Its binding to importin β releases the auto-inhibitory domain of importin α, which in turn detaches from the NLS carrying protein cargo (Fig. 1c). Most importantly, only RanGTP, and not RanGDP, can bind to importin β. This is because the importin β binding domain of Ran is only accessible when Ran is bound to GTP (Fig. 1d).
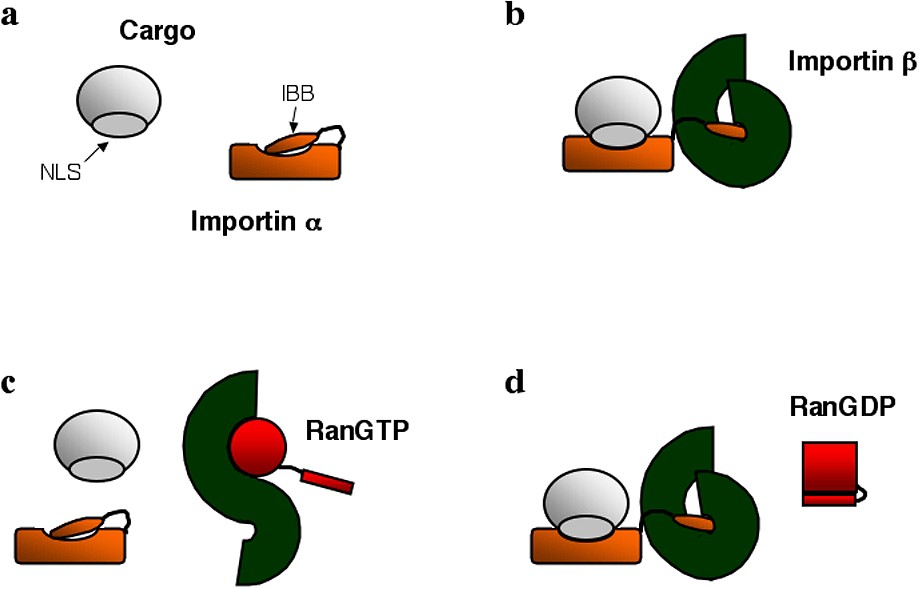
Schematic representation of various soluble factors involved in nuclear import: import cargo carrying a nuclear localization signal (NLS), importin α with its importin β binding domain (IBB), importin β, RanGTP and RanGDP. Panels (a), (b), (c) and (d) highlight the possible interactions among these proteins.
In this article, we report our efforts to apply fluorescence correlation spectroscopy (FCS) [19–22] to the study of the formation and dissociation of transport cargoes. These experiments were carried out in vitro with purified proteins and fluorescently labeled peptides carrying an NLS sequence. The advantage of using FCS over other techniques, for example, plate-based biosensors, is that the actual solution interactions of the proteins are probed. Our study confirms that NLS carrying peptides and importin α can bind, but only in the presence of importin β, and only when the peptide carries the correct NLS sequence. It also confirms that RanGTP, but not RanGDP, is able to dissociate the cargo complex. In addition, we show that RanGTP binds preferentially to importin α/importin β complexes that do not carry a transport cargo. This finding raises the interesting question of whether importin β complexes may be dissociated preferentially by RanGTP according to their target.
2 Materials and methods
2.1 Fluorescent peptides
The sequences of the two peptides used in this study were CTPPKKKRKV and CTPVKRKKKP. The first peptide, referred to as NLS peptide in the following, contains the nuclear localization signal corresponding to residues 126–132 on the SV40 large T-antigen, PKKKRKV. This NLS is connected to a C-terminal cysteine by a linker sequence, TP. The second peptide, referred to as bNLS peptide in the following, is similar to the first one, except for the fact that the NLS sequence has been inverted. As a result, both peptides have the same overall electrostatic properties, but only the first one contains the proper amino acid sequence for identification as an import cargo. Both peptides were synthesized in the lab of Dr. Mati Fridkin, and purified by preparative thin-layer chromatography. MALDI-TOF mass spectrometry showed that the purified peptides had the expected 1.18-kDa molecular weight. They were subsequently linked to an Alexa dye (Alexa Fluor 546 C5 maleimide, Molecular Probes) through maleimide chemistry. Briefly, 0.6 mM of peptide was allowed to react with a 1.3-fold excess of Alexa dye in a phosphate buffer (NaH2PO4, 50 mM, pH=7.1, adjusted with NaOH) for 2 h at room temperature and then overnight at 4 °C, while rotating the solution end-to-end continuously. The crude reaction mixture was purified in a size exclusion column (P-2 Bio-Gel, from Bio-Rad) with an exclusion limit of 1.8 kDa. The reaction mixture was eluted with ammonium hydrogen carbonate (0.1 M) at 4 °C. The collected fractions were analyzed by thin-layer chromatography. The product was lyophilized and re-dissolved in water twice to remove salt. Analysis by mass-spectrometry confirmed that the product had the expected 2.2-kDa molecular weight.
2.2 Purified proteins
Purified human importin α and human importin β were a kind gift of Dr. Dirk Görlich. Purified RanQ69L was a kind gift of Dr. Renat Nevo. RanQ69L is a mutant of human Ran, which inhibits GTP hydrolysis [23]. RanQ69L-GTP is very unlikely to hydrolyze into RanQ69L-GDP. The method used for loading the protein with either GDP or GTP is described in detail in [24]. The purity of all proteins was checked by SDS–PAGE, and the concentrations of the stock solutions were measured by absorption at 280 nm. All the titrations presented here were carried out in PBS buffer (pH=7.4) containing 0.1% digitonin and
2.3 Fluorescence correlation spectroscopy
A home-built apparatus, described elsewhere [25], was used to perform FCS experiments. Briefly, fluorescence was excited by a 543.5-nm HeNe laser focused in the sample by an oil objective (Zeiss, Achrostigmat,
Before each experiment, the detection volume was first calibrated using the diffusion of Rhodamine 610, purchased from Exciton. The autocorrelation curves obtained from diffusion of the dye were analyzed using the expression [26]:
| (1) |
Eq. (1) was also used to account for the autocorrelation curves obtained from the diffusion of the peptides in the absence of the soluble transport factors importin α and importin β. On the contrary, the autocorrelation curves obtained from the diffusion of the peptides in presence of the transport factors were analyzed assuming a two-component model accounting for the presence of cargo complexes [28]:
| (2) |
| (3) |
| (4) |
| (5) |
2.4 Kinetic analysis
For titration curves involving the association of a receptor (typically the importin α/β heterodimer, noted AB) with a ligand (typically the NLS peptide, noted P), the reaction was described by a simple equilibrium:
| (6) |
| (7) |
| (8) |
However, if there are two different types of complexes in solution (in our case importin α/β heterodimers on their own and actual cargo complexes, i.e. importin α/β heterodimers bound to a NLS peptide), noted Cαβ and CNLSαβ, we should consider the possibility that Ran binds with different affinities to the two complexes. If this is the case we have two coupled reaction equations:
If
| (9) |
3 Results
Our earliest attempts at titrating solutions of peptides with different purified proteins showed that, as the total protein concentration in the solution was increased, and although the amount of peptide per sample was kept constant, the concentration of peptides detected in solution increased. The increase in soluble peptide concentration was indicated both by an increase in the total average intensity and by an increase in the average number
To avoid as much as possible the loss of proteins and peptides to glass and polymer surfaces while handling the solutions and taking the measurements, we then supplemented the solution with ovalbumin. We tested increasing concentrations up to
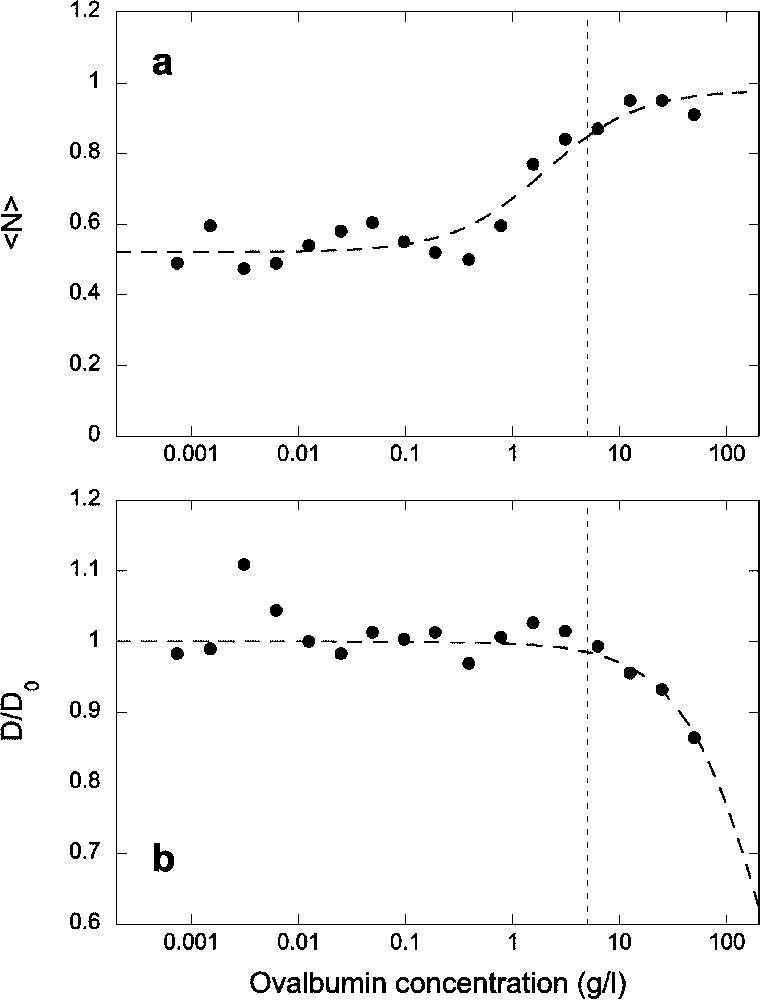
(a) Average number of fluorescent Alexa-NLS peptides in the detection volume and (b) normalized diffusion coefficient of the peptide, as a function of ovalbumin concentration. All data have been obtained for a constant concentration of the peptide c=5 nM introduced in the sample. Dashed lines are guide for the eyes. The vertical dotted lines indicate the concentration of ovalbumin chosen for subsequent experiments.
We first measured the binding of the importin α/β heterodimer to the Alexa-NLS peptide. The α/β complex was allowed to form in an equimolar 20 μM solution of importin α and importin β. Different volumes of this solution were then mixed with a second solution containing the peptide. The final concentration of peptides in these mixed solutions was
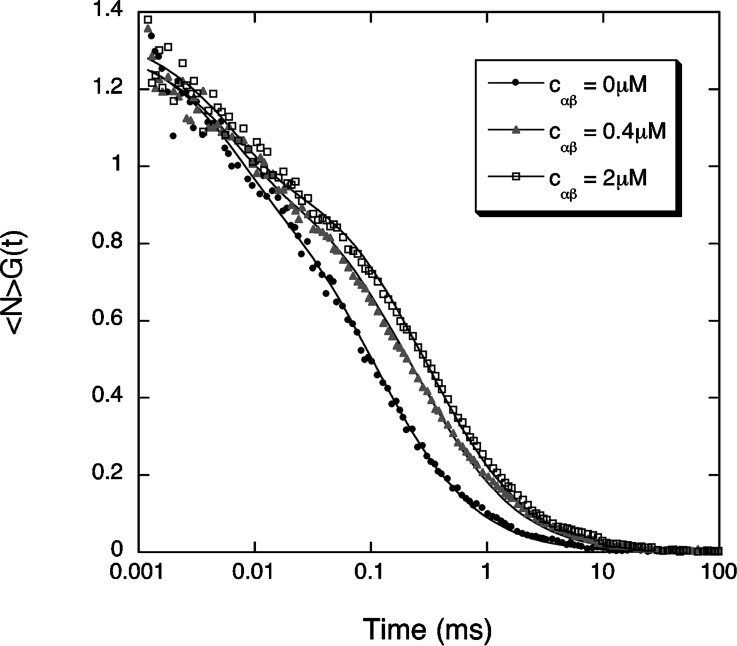
Example of three autocorrelation curves measured for the fluorescent NLS peptide in the absence (black dots) or in the presence of an equimolar concentration
Example of three autocorrelation curves measured for the fluorescent NLS peptide in the absence (black dots) or in the presence of an equimolar concentration
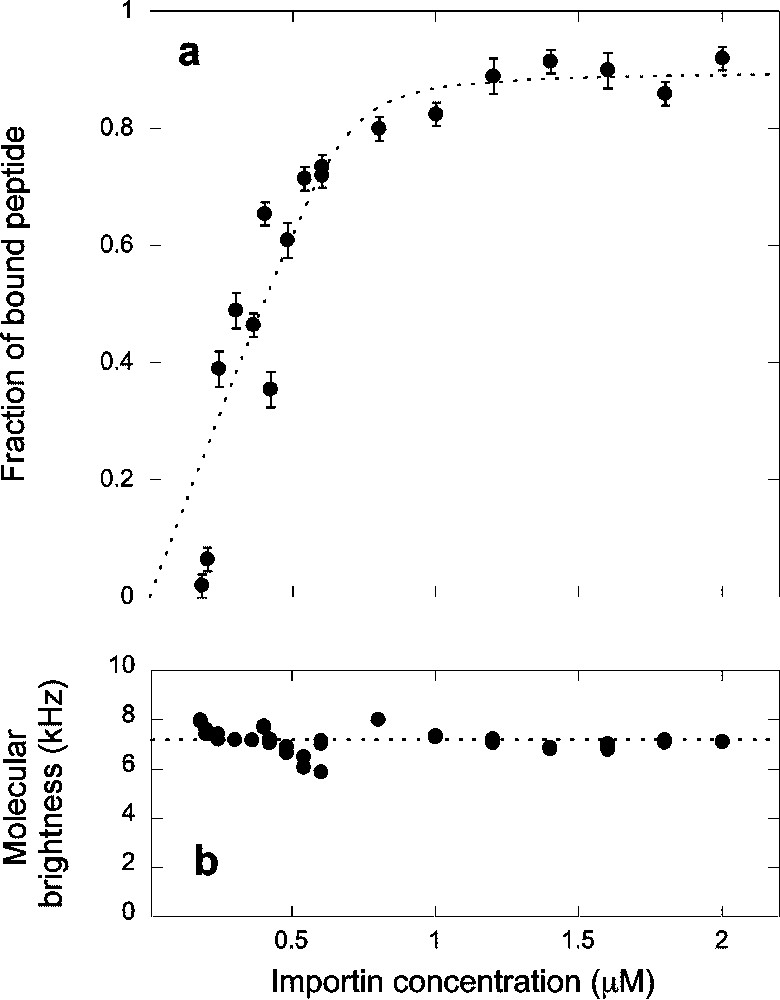
(a) Titration curve showing the fraction of peptide bound to the complex formed by importin α and importin β and (b) apparent fluorescence per molecule calculated as explained in the text, as a function of the concentration
(a) Titration curve showing the fraction of peptide bound to the complex formed by importin α and importin β and (b) apparent fluorescence per molecule calculated as explained in the text, as a function of the concentration Lire la suite
The binding curve in Fig. 4a was analyzed using the simple binding model leading to Eq. (7). The fit of the data presented in Fig. 4a with this model gave
To validate the result of this experiment, several different titrations were performed as controls. First we checked that the α/β complex was binding specifically to the NLS sequence of the Alexa-NLS peptide, by repeating the same titration using the Alexa-bNLS peptide. Although some evidence of binding at high importin concentration was detected, the dissociation constant
| Peptide | Ligand |
Dissociation constant
|
| Alexa-NLS | α/β heterodimer | 12±3 nM |
| Alexa-bNLS | α/β heterodimer | >100 nM |
| Alexa-NLS | Importin α | >1 μM |
| Alexa-NLS | α/β heterodimer, RanGDP present | 2±10 nM |
| Alexa-NLS | α/β heterodimer, RanGTP present | >1 μM |
| α/β heterodimer | RanGTP | <0.2 nM |
| Alexa-NLS/α/β heterodimer | RanGTP | 18±16 nM |
We next studied the dissociation of the cargo complex by RanGTP. For this, we titrated solutions containing a concentration
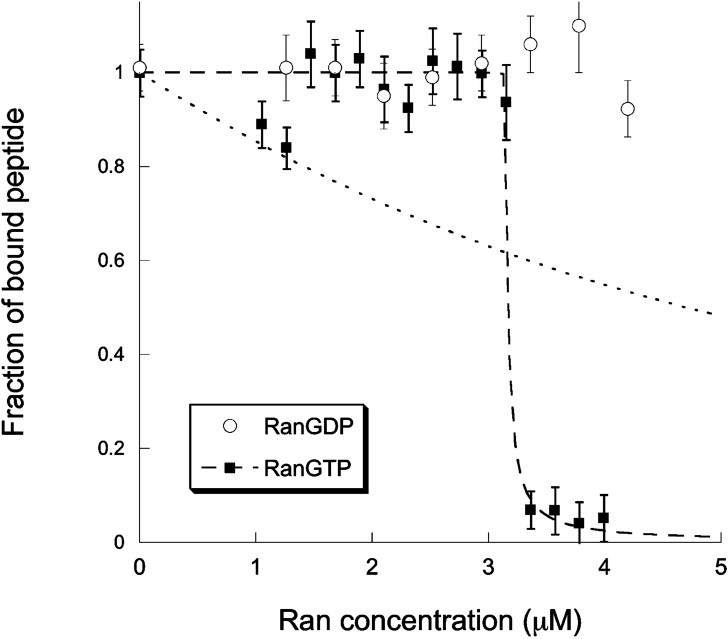
Titration curves showing the fraction of Alexa-NLS bound to a cargo complex as a function of the introduced concentration of RanGTP (filled squares) or RanGDP (empty circles). The lines represent fits of the RanGTP titration curve using either Eq. (8) (dotted line) or Eq. (9) (dashed line). This second fit gives
4 Discussion
Fluorescence correlation spectroscopy is a powerful method that allows detection of binding of fluorescent ligands to receptors directly in solution. The diffusion coefficient of the receptor just needs to be 1.6 times larger than that of the ligand [27]. Since FCS is very well suited for working with nanomolar concentrations of reactants, it should be an ideal method to measure interactions between biomolecules, for which binding is often very tight, with dissociation constants in the nM range. However, when trying to determine equilibrium constants for reactions involving soluble components, surface adsorption is a critical problem. It can severely reduce the concentration of the reactants in solution, introducing several unknown variables. In particular, the negative charge density of glass surfaces, acquired through the dissociation of silanol groups, is an issue for the study of small concentration of positively charged or hydrophobic peptides. We were able to alleviate this problem by using ovalbumin to crowd the solution. For the highly positively charged peptides we studied, an ovalbumin concentration of at least 1 g l−1 was required to begin to displace the peptides from the glass surfaces. At 5 g l−1, the ovalbumin was able to compete efficiently against the peptide for surface adsorption, and above 10 g l−1 all the peptides were displaced from the surfaces. On the other hand, above a 5-g l−1 concentration of ovalbumin, we began to see an influence of the ovalbumin on the peptide diffusion, with a diffusion coefficient reduced by more than 10% at 10 g l−1. This is probably due to an increase in the effective viscosity of the solution, though it is also possible that at these high concentrations the peptide starts to interact with the protein, or that inhomogeneities form in the solution due to depletion interactions. For this reason, we chose not to use ovalbumin concentration above 5 g l−1. The fact that the molecular brightness of the fluorophore did not change when increasing the ovalbumin concentration showed that up to 50 g l−1 the optical index mismatch created by the addition of protein is not large enough to cause strong optical aberrations.
Because of adsorption on the sample chamber surfaces, the precision with which we could evaluate the concentration of the reactants, and consequently extract binding constants, proved to be limited. Relative measurements, on the other hand, should not be strongly affected by such uncertainties. While the peptide concentration in solution could be monitored through the FCS measurement, the concentration of the purified proteins (importin α, importin β, and Ran) had to be estimated. The fit to the data in Figs. 4a and 5 give sensible results only if the protein concentrations are allowed to float. This emphasizes the usefulness of cross-correlation spectroscopy, where the absolute concentration of both receptor and ligand can be monitored, and where there is no need for one reactant to be much larger in size than the other. Measurement of dissociation constants larger than 1 μM was problematic, as the concentration of ligand required in order to achieve full binding is higher than what could be obtained. In those cases, the values measured have a large uncertainty associated with them.
Our titration experiments are all in good agreement with the equilibrium constants measurements determined by other techniques [15,16,29]. For the interaction of the SV40 large T-antigen NLS with the importin α/β heterodimer, we measure a dissociation constant
Overall, our experiments with RanGTP and RanGDP are also in agreement with the accepted model of nuclear transport, which is based on the notion that the gradient of RanGTP across the nuclear membrane drives vectorial transport by dissociating cargo complexes once they reach the nucleus. Our titration shows that indeed RanGTP is able to dissociate cargo complexes and to release the NLS carrying cargo, while RanGDP has no noticeable effect on the cargo complexes. Moreover, we show that RanGTP has a different affinity for importin β when it is part of a α/β heterodimer than when it is part of a NLS/α/β cargo complex. It has been assumed previously that the affinity of RanGTP for a complete cargo complex might be reduced about five times compared to its affinity for the α/β heterodimer alone [30]. Our measurements validate this hypothesis. Although the binding constants we measured should be taken as estimates rather than exact values, it emerges from the data presented in Fig. 5 that the affinity of RanGTP for importin β when part of an α/β heterodimer is at least 10 times larger than for importin β when it is part of a complete NLS/α/β cargo complex. The effect we observe may be due to a much tighter binding between importin α and importin β in presence of a NLS carrying cargo, making it more difficult for RanGTP to take advantage of transient conformational changes of the complex to bind to importin β. Indeed, the rate constant for dissociation of the α/β heterodimer has been measured to be as high as 0.02 s−1 [30], indicating that this complex might undergo structural fluctuations leading to conformational changes partially exposing the Ran binding domain of importin β. There is no information available about the dissociation rate of importin β from importin α when the latter is bound to a NLS. Our experiments could suggest that this rate constant is at least 10 times smaller than in the absence of the NLS sequence.
The fact that the dissociation of cargo complexes depends on whether or not they are actually loaded with a cargo might have interesting consequences in nucleocytoplasmic transport. It could mean that different cargoes, which bind to importin α with different affinities [29], will be dissociated by RanGTP in a different order. If the rate-limiting step for the import of a specific cargo is the dissociation of the cargo complex by RanGTP, then the kinetics of import might be very dependent on the type of NLS carried by the cargo. In the larger context of the role of the soluble nuclear import receptors as control proteins in various mitotic processes, the possibility of a preferential release of importin β when it binds to different targets is also quite interesting. It could explain how the very different molecular switch functions of the importin β/RanGTP pair may be orchestrated by the cell. It has been proposed that this difficult coordination could be achieved by the presence of other (still remaining to be identified) regulators of importin β, in addition to RanGTP [7]. However, both importin β and RanGTP are very abundant, and it is unlikely that a new regulator of importin β, if it is discovered, could be as dominant as Ran. Our results suggest a different explanation, namely that RanGTP itself, through its preferential affinity for importin β when bound to specific cargoes, is able to regulate which targets of the importin α/β heterodimer it will release.
Acknowledgements
We thank Dr. D. Görlich and Dr. R. Nevo for providing us with purified proteins, and Dr. M. Peretz for expert technical assistance. This work was supported in part by a grant from the Israel Science Foundation – The Charles H. Revson Foundation, and by the Gerhardt M.J. Schmidt Minerva Center for Supramolecular Architecture. C.F. was supported partly by a Marie-Curie research fellowship, and partly by the Natural Sciences and Engineering Research Council of Canada through its CRC program.
Vous devez vous connecter pour continuer.
S'authentifier