1 Introduction
HIV genome flexibility is characterized by a high frequency of spontaneous mutations and a variety of rearrangements. The error-prone replication by HIV reverse transcriptase is generally held responsible for the impressive fraction of defective viruses observed in productively infected lymphocytes. A variety of other mechanisms can contribute to generate modifications in the HIV genomes by restabilizing the viral information under new forms, among them recombination phenomena now thought to be mainly responsible for HIV genomic flexibility throughout an infection process [1–4]. This raised the notion of ‘mosaic viruses’ composed of parts inherited from divergent entire or partial viral genomes that could be present in the cells when the replication steps occur [5–7].
As lentiviral genomes are composed of an alternation of long variable and short conserved sequences, this appeared to be a characteristic of each part of the genomes organized as a succession of segments that can evolve differently and independently. The sequence conservation can be considered as the maintenance of either the general function of a protein or a potential precise function associated with a nucleotide segment (e.g., restriction or binding site). The extended nucleotide variability allowed by natural selection could lead to the evolution or to the disappearance of a function. In view of this analysis, the short conserved sequences may play crucial roles both in viral ontology and viral divergence independently of gene products functions (e.g., regulatory or enzymatic processes) as some of these sequences overlap genes. They could correspond to recombinogenic sequences important to understand lentiviral genomic flexibility.
Studies of the HIV genetic variability required computerized methods to investigate genomic divergences [8–11]. A Recombinant Identification Program was applied to the HIV-1 gag and env coding regions and allowed to determine putative large recombination segments – thus delimiting ‘recombination cassettes’ – and to create phylogenetic trees [12]. However, theses trees highly differed when the currently used computation methods were applied to either the gag, pol, or env genes, and when reference genome in the same population was changed [6,7,12–14]. The situation was made more complex when fragment analysis of a single gene showed divergent phylogenetic trees for each studied fragment [5–7,14–16], this being due to the independent evolution of each gene. In lentiviral genomes, the programs have rarely revealed precise recombinogenic segments, but rather computerized plots or large domains possibly implied in the recombination process [10,11,17].
Comparison of whole genomes of related ‘species’ (with the meaning of taxons) is currently considered to identify the genomic organization and functionality without prior biological characterization [8,18]. In our global approach concerning complete genomes in all situations, we carried out the detection of Conserved Lentiviral Sequences (CLSs), their precise location being harmonized thanks to the use of a single MMy 1® sequence starting reference (see Section 2). The analysis was made on genomes belonging to mammal lentiviruses, negative control viruses and random-generated genome-like sequences. This scan of the DNA lentiviral sequences for conserved stretches allowed to identify 28 CLSs mainly situated in gag, pol and, in a minor proportion, env structural genes. A few of them were also noticed in the LTRs and the regulatory genes. A large part (82%) of CLSs located in HIV-1s is situated in currently described recombination segments where they might form recombinogenic hot spots [6,7,13–15,19–36]. The similar position of each sequence in the different viral families led to establish the notion of ‘lentiviral’ and ‘retroviral self’ that was defined by the specificity of the sequences for restricted viral families.
2 Methods
2.1 Studied sequences
The viral genomes were retrieved from the Genbank database and their loci are listed in Appendix A. Immunodeficiency lentiviral genomes correspond to 171 human viruses (155 HIV-1s and 16 HIV-2s), 33 simian viruses (3 CPZ, 9 AGM, 8 Macaque, 2 Mandrill, 10 Sooty Mangabey, 1 Sykes' monkey viruses) and 33 non-primate mammal viruses (2 bovine, 2 caprine, 11 equine, 9 feline, 3 ovine and 6 ovine/caprine viruses). To test CLS specificity, three kinds of negative controls were examined. First, non lentiviral yet retroviral genomes were screened (5 spumaviruses, 25 oncornaviruses and 4 D-particle-forming simian viruses). Then, a set of human, animal and vegetal viruses was tested: 4 herpes viruses, 47 human and animal viruses (1 adenovirus, 6 coronaviruses, 2 filoviruses, 12 flaviviruses, 13 parvoviruses, 10 picornaviruses and 3 rhabdoviruses) and 18 vegetal viruses (5 geminiviruses, 2 potyviruses, 5 tobamoviruses, 1 tombusvirus, 3 tymoviruses and 2 necroviruses). Finally, 50 10-kilobase-DNA-like structures were randomly generated with a computer in order to eliminate any bias due to the possible subsequent biological role.
2.2 Determination of conserved sequences
Using a previously published program [8,37], a general analysis was carried out to identify the highly conserved sequences common to all or a maximum of lentiviral genomes. We first established the length of sub-sequences with the following trial/error method selecting parts of genomes. When the length was inferior to 10 nucleotides, numerous sub-sequences were found in every genome, including the control ones. When it was superior to 50 nucleotides, very few sub-sequences were found. The correct length was determined when it corresponded to that of sub-sequences common to most of the immunodeficiency viral genomes in either all or certain viral families. A similar approach was used for the determination of the number of accepted transitions. The relationship between both parameters led to an optimized choice of a 15-nucleotide-long sequence with a maximum of three admitted transitions. All the sequences showing such a determined length were tested and positioned in genomes thanks to the Expasy program available on Internet. Some of them overlapped and created longer conserved domains. Sequences present in most lentiviral genomes and checked not to be found in negative controls were selected. The number of accepted transitions was retained according to their lentiviral specificity to allow a variability of 15 to 20%. In some cases the limit of 25% was retained, which is thought to delimit the generally admitted jump from one family to another and might fit with a possible biological significance.
2.3 MMy 1® as reference for sequence position
The genomes supplied by the databases show variable lengths because LTRs are reported with different lengths. Thus, as the first considered nucleotides at the 5′ extremity vary from one genome to another, the rough detection of the sub-sequences gave heterogeneous localizations. Another approach could use the real starting ATG (as given in the databases) of the coding sequences as the initial nucleotide, but this was too complex because of the frequent presence of several other ATGs. To sharpen and to harmonize the location of CLSs, we calculated their relative positions as compared to that of one of them chosen as the reference sequence (position +1). This sequence – MMy 1® – was previously described for PCR purpose [38,39] and corresponds to the beginning of the PBS (reverse transcription primer binding site) at the 5′ extremity of the genomes. It is situated, for example, between nucleotides 182 and 199 on the HIV-1 BRU/LAI genome reported in Genbank. MMy 1® was found in HIV-1, HIV-2, AGM, macaque, CPZ, sooty mangabey, mandrill, equine and feline lentiviruses. When two LTRs were present in the genomic banks (HIV-1, HIV-2 and equine viruses), a second MMy 1® sequence detected at the 3′ extremity of the genomes was not considered in the analysis. In the Sykes' monkey, bovine, caprine, ovine and ovine/caprine viruses lacking MMy 1®, the indicated numbers corresponding to CLS locations were determined as crude positions directly detected on the genomes.
3 Results
When investigated on 237 complete viral genomes, 28 CLSs were detected, six of them being the MMy® ones that have been previously used as PCR primers for the early detection of possible HIV infection in highly exposed population [38,39]. These sequences together with some new determined CLSs classified from CLS1 to CLS 22 are shown in Table 1. The relationship between precise positions of CLSs and genomic organization of the viral families are depicted in Figs. 1 to 3. The CLS characteristics together with their specific gene locations shown in Tables 1–3 were divided into categories according to their lentiviral specificity.
Characteristics of CLSs detected in lentiviral genomes
| Seq. names | Nucleotidic sequences | Mersa | Max. trans.b | Viral genomesc |
| MMy 1 | tggcgcccgaacagggac | 18 | 3;17% | all HIV-1s, HIV-2s, AGMs, CPZs, mac., sooty, mnd., equ., fel. |
| MMy 3 | taaaacatttagtatgggca | 20 | 3;15% | all HIV-2s, AGMs, mac., CPZs and 152/155 HIV-1s, 9/10 sooty, 1/2 mnd. |
| MMy | catcaagcagctatgcaaat | 20 | 3;15% | all HIV-2s, mac., sooty and 146/155 HIV-1s, 1/9 AGM, 2/3 CPZs |
| MMy 28 | agggctgttggaaatgtgg | 19 | 3;16% | all HIV-2s and 154/155 HIV-1s, 7/8 mac., 8/10 sooty, 1/3 CPZ |
| MMy 31 | catgggtaccagcacacaaagg | 22 | 4;18% | all HIV-2s, AGMs, CPZs, mac., sooty, mnd., equ., Sykes' monkey and 154/155 HIV-1s, 2/3 ov. |
| MMy 32 | tggaaaggggaaggagcagt | 20 | 3–4;15–20% | all HIV-1s, HIV-2s, AGMs, mac., sooty, CPZs, Sykes' monkeys, mnd., cap., ov. |
| CLS 1 | atagaagcagaagttat | 17 | 3;18% | all HIV-1s, HIV-2s, AGMs, CPZs, mac., sooty, mnd. and 1/2 cap., 3/9 fel., 2/3 ov. |
| CLS 2 | tttttaaaagaaaagggg | 18 | 3;17% | all CPZs, mnd., sooty, equ., Sykes' monkey and 153/155 HIV-1s, 15/16 HIV-2s, 8/9 AGMs, 7/8 mac. |
| CLS 3 | tagatacaggagctgatg | 18 | 3;17% | all AGMs, CPZs, Sykes' monkey, bov., equ. and 153/155 HIV-1s, 14/16 HIV-2s, 7/8 mac., 9/10 sooty, 1/2 mnd., 7/9 fel., 2/6 ov./cap. |
| CLS 4 | acaaggaccaaaggaacc | 18 | 3–4;17–22% | all HIV-1s, HIV-2s, mac., sooty, mnd., equ. and 7/9 AGMs, 1/3 CPZ, 1/2 bov., 1/2 cap., 6/9 fel., 3/6 ov./cap./ |
| CLS 5 | catgggtaaaagtagtaga | 19 | 3;16% | all CPZs, ov./cap. and 154/155 HIV-1s, 15/16 HIV-2s, 4/9 AGMs, 3/8 mac., 4/10 sooty, 1/2 mnd., 1/2 cap., 2/3 ov. |
| CLS 6 | aaattttcgggtctattacagagaaggcagagatc | 35 | 6;17% | all HIV-2s, AGMs, CPZs, mac., sooty, mnd. and 152/155 HIV-1s |
| CLS 7 | gctacttccctgattg | 16 | 3;19% | all HIV-1s and 2/3 CPZs |
| CLS 8 | ccacctggattcctga | 16 | 2–3;12–19% | all HIV-1s, CPZs and 1/10 sooty |
| CLS 9 | agtactaaatggagaaaa | 18 | 2–3;11–17% | all CPZs and 152/155 HIV-1s |
| CLS 10 | aaaaataaccacagaaagc | 19 | 4;22% | all CPZs, cap. and 141/155 HIV-1s, 1/16 HIV-2, 1/2 mnd., 1/10 sooty |
| CLS 11 | caggggaaagaatagtaga | 19 | 3;16% | all CPZs, mnd., ov./cap. and 153/155 HIV-1s, 2/3 ov. |
| CLS 12 | acaaaaaacatcagaaagaacc | 22 | 3;14% | 153/155 HIV-1s, 1/3 CPZ, 1/2 mnd. |
| CLS 13 | aataaaacaaattataaacatg | 22 | 4;18% | 143/155 HIV-1s, 1/2 cap. |
| CLS 14 | acaaaagtaagaaaaaagcacagcaagc | 28 | 1–7;4–25% | 125/155 HIV-1s |
| CLS 15 | aaactaaagaattacaaaaacaaattacaaaaatt | 35 | 6;17% | 134/155 HIV-1s, 1/3 CPZ |
| CLS 16 | attttaaaagaaggggag | 18 | 3;17% | all Sykes' monkey, mnd. and 144/155 HIV-1s, 12/16 HIV-2s, 1/3 CPZ, 5/8 mac., 4/10 sooty, 1/9 AGM, 1/9 fel. |
| CLS 17 | tccatatccacaaggacc | 18 | 3;17% | 15/16 HIV-2s, 5/8 mac., 3/10 sooty, 1/9 AGM, 1/2 cap., 1/155 HIV-1 |
| CLS 18 | agtaccaacagggtcaga | 18 | 3;17% | all HIV-2s, AGMs, mac. and 8/10 sooty, 1/2 mnd., 1/9 fel. |
| CLS 19 | agcaccacctagcgggag | 18 | 3;17% | all mac. and 14/16 HIV-2s, 8/10 sooty |
| CLS 20 | agcaacagctgttggacg | 18 | 3;17% | all HIV-2s, mac., sooty and 1/9 fel. |
| CLS 21 | cctcctcctcctccccctccagg | 23 | 3;13% | all mac., sooty and 15/16 HIV-2s |
| CLS 22 | tgaaaaacctcaatgccc | 18 | 3;17% | all AGMs, Sykes' monkey and 1/2 mnd |
a Mer-length of the sequences.
b Maximum of admitted transitions followed by the percentage of variation they represent.
c Ratio of genomes in which the tested sequence was found for each viral family.

CLSs localization and genomic organization of HIV-1 and HIV-2. The numbers represent the relative positions of the detected CLS versus that of the MMy 1® (see Section 2). The reference organization of HIV-1 and HIV-2 genomes was represented using HIVBRUCG and HIV-2CAM2 viruses, respectively. Occn.: CLS occasionally found on less than 10% of the studied genomes; occp.: CLS found at an occasional position.

CLSs localization and genomic organization of AGM and Macaque viruses. The numbers represent the relative positions of the detected CLS versus that of the MMy 1® (see Section 2). The reference organization of AGM and Macaques viral genomes was represented using SIVAGM155 and SIU72748 viruses, respectively. Occp.: CLS found at an occasional position.
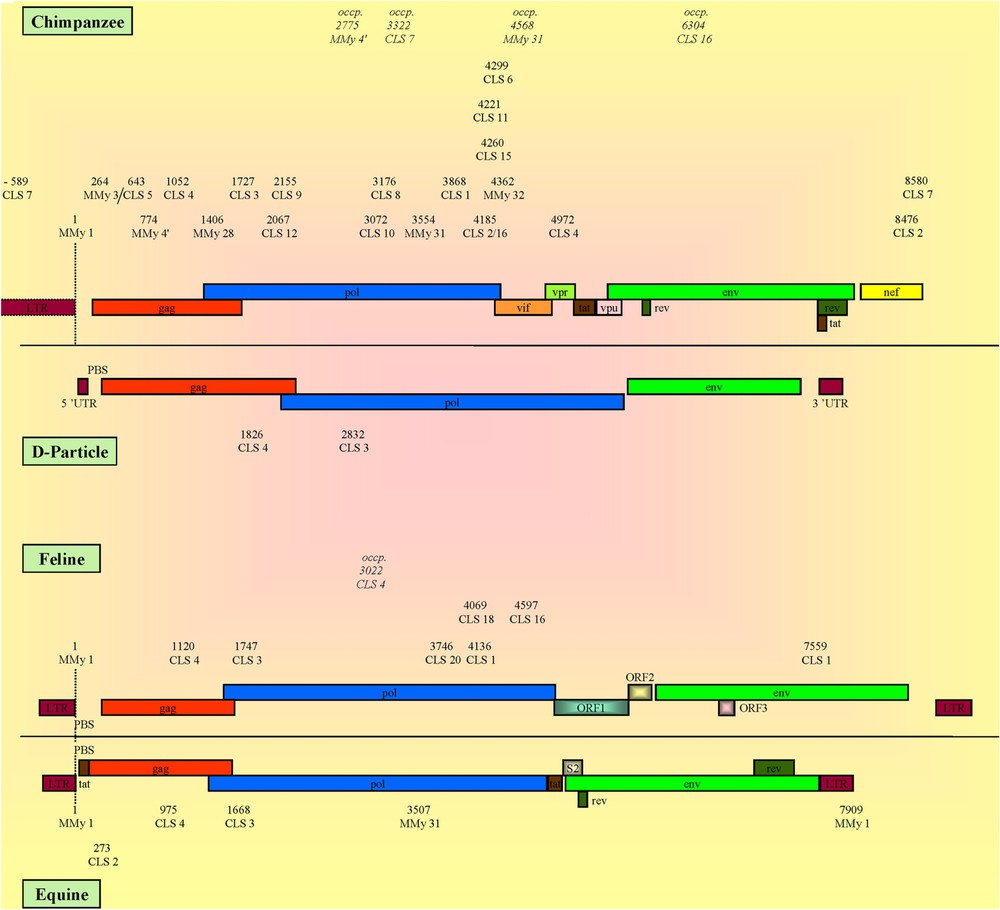
CLSs localization and genomic organization of CPZ, D-particle-forming viruses, feline and equine viruses. The numbers represent the relative positions of the detected CLS calculated versus that of the MMy 1®, except for D-particle-forming viruses that lack MMy 1®, whose numbers correspond to the crude positions of the CLSs directly referenced from the genomes (see Section 2). The reference organization of CPZ, D-particle-forming viruses, feline and equine viruses was represented using: AF103818, AF033815, FIVZ1 and AF247394 viruses, respectively. Occp.: CLS found at an occasional position.
Classification of CLSs detection according to primate lentiviral families
| HIV-1 | HIV-2 | AGM | Macaques | Sooty M. | CPZ. | Mandrill | Sykes' monkey | |||||||||
| MMy 1 | ●a | PBS – U5b | ● | PBS – U5 | ● | PBS | ● | PBS | ● | PBS | ● | PBS | ● | PBS | ||
| MMy 3 | ● | gag (p17) | ● | gag | ● | gag | ● | gag | ● | gag | ● | gag | ■ | gag | ||
| MMy | ● | gag (p24) | ● | gag | ■ | gag – pol | ● | gag | ● | gag | ■ | gag / pol | ||||
| MMy 28 | ● | gag (p7) | ● | gag | ■ | gag | ■ | gag | ■ | gag / pol | ||||||
| MMy 31 | ● | pol (p15) | ● | pol | ● | pol | ● | pol | ● | pol | ● | pol / vif | ● | pol | ● | pol |
| MMy 32 | ● | pol (p31) | ● | pol | ● | pol | ● | pol | ● | pol | ● | vif | ● | pol | ● | vif |
| CLS 1 | ● | pol (p31) / | ● | pol / vpr | ● | pol / pol | ● | pol | ● | pol / env | ● | pol | ● | pol / vif | ||
| pol (p31) | ||||||||||||||||
| CLS 2 | ● | pol (p31) / nef | ● | pol / nef | ■ | nef | ■ | nef | ● | nef | ● | pol / nef | ● | pol / nef | ● | vif |
| CLS 3 | ● | gag (p6) – | ■ | pol | ● | pol | ■ | pol | ● | pol | ● | pol | ■ | pol | ● | pol |
| pol (p10) | ||||||||||||||||
| CLS 4 | ● | gag (p24) | ● | gag / env | ■ | gag | ● | gag | ● | gag | ■ | gag / vpr | ● | gag | ||
| CLS 5 | ● | gag (p24) | ● | gag | ■ | gag | ■ | gag | ■ | gag | ● | gag | ■ | gag | ||
| CLS 6 | ● | pol (p31) | ● | pol | ● | pol | ● | pol | ● | pol | ● | pol / vif | ● | pol | ||
| CLS 7 | ● | nef / U3 | ■ | LTR / nef / pol | ||||||||||||
| CLS 8 | ● | pol (p51) | ■ | pol | ● | pol | ||||||||||
| CLS 9 | ● | pol (p51) | ● | pol | ||||||||||||
| CLS 10 | ● | gag (p17) / | ○ | env | ■ | env | ● | pol | ■ | pol | ||||||
| pol (p51) | ||||||||||||||||
| CLS 11 | ● | pol (p31) / | ● | pol | ● | pol | ||||||||||
| env (gp120–V3) | ||||||||||||||||
| CLS 12 | ● | pol (p51) | ■ | pol | ■ | pol | ||||||||||
| CLS 13 | ● | pol (p31) / | ||||||||||||||
| env (gp120–V4) | ||||||||||||||||
| CLS 14 | ■c | gag (p17) / vpu | ||||||||||||||
| CLS 15 | ■ | pol (p31) | ■ | pol | ||||||||||||
| CLS 16 | ● | pol (p31) | ■ | pol | ■ | pol | ■ | pol | ■ | pol | ■ | pol / env | ■ | pol | ● | vif |
| CLS 17 | ○ | vif | ● | env | ■ | rev | ■ | env | ■ | tat – rev – env | ||||||
| CLS 18 | ● | gag / gag / pol | ● | gag / gag | ● | gag / gag | ■ | gag | ■ | gag | ||||||
| CLS 19 | ■ | gag | ● | gag | ■ | gag | ||||||||||
| CLS 20 | ● | env | ● | env | ● | env | ||||||||||
| CLS 21 | ● | vpx | ● | vpx | ● | vpx | ||||||||||
| CLS 22 | ● | env | ■ | gag | ● | tat |
a Detection degree of the tested CLS in the genomes: (●) at least 90%; (■) comprised between 10% and 89%; (○) less than 10%.
b Gene location of the CLSs: genes separated by ( / ) when CLS was detected on each of the two genes; genes separated by (–) when CLS overlapped the 2 genes; gene indicated in italics when CLS was present at an occasional position.
c A gradual detection of CLS 14 was observed when the admitted transitions in this sequence varied from 1 (66%) to 7 (85%).
Classification of CLSs detection according to non-primate mammal lentiviral families
| HIV-1 | HIV-2 | Bovine | Equine | Feline | Caprine | Ovine | Ov./Cap. | |||||||||
| MMy 1 | ●a | PBS – U5b | ● | PBS – U5 | ● | PBS – LTR | ● | PBS | ||||||||
| MMy 31 | ● | pol (p15) | ● | pol | ● | pol | ■ | pol | ||||||||
| MMy 32 | ● | pol (p31) | ● | pol | ● | pol | ● | pol | ||||||||
| CLS 1 | ● | pol (p31) / | ● | pol / vpr | ■ | pol / env | ■ | pol | ■ | env | ||||||
| pol (p31) | ||||||||||||||||
| CLS 2 | ● | pol (p31) / nef | ● | pol / nef | ● | gag | ||||||||||
| CLS 3 | ● | gag (p6) – | ○ | pol | ● | pol | ● | pol | ■ | pol | ■ | pol | ||||
| pol (p10) | ||||||||||||||||
| CLS 4 | ● | gag (p24) | ● | gag / env | ■ | gag / pol | ● | gag | ■ | gag / pol | ■ | env | ■ | pol | ||
| CLS 5 | ● | gag (p24) | ● | gag | ■ | pol | ■ | pol | ● | pol | ||||||
| CLS 10 | ● | gag / pol (p51) | ○ | env | ● | vif | ||||||||||
| CLS 11 | ● | pol (p31) / | ■ | env | ● | env | ||||||||||
| env (gp120-V3) | ||||||||||||||||
| CLS 13 | ● | pol (p31) / | ■ | n.a.c | ||||||||||||
| env (gp120-V4) | ||||||||||||||||
| CLS 16 | ● | pol (p31) | ■ | pol | ■ | pol | ||||||||||
| CLS 17 | ○ | vif | ● | env | ■ | pol | ||||||||||
| CLS 18 | ● | gag / gag / pol | ■ | pol | ||||||||||||
| CLS 20 | ● | env | ■ | pol |
a Detection degree of the tested CLS in the genomes: (●) at least 90%; (■) comprised between 10% and 89%; (○) less than 10%.
b Gene location of the CLSs: genes separated by ( / ) when CLS was detected on each of the two genes; genes separated by (–) when CLS overlapped the 2 genes; gene indicated in italics when CLS was present at an occasional position.
c n.a.: not attributed.
3.1 Sequences common to a maximum of HIV-1, HIV-2 and SIV isolates
These sequences included all the MMy ones, CLS 1 to 6 and CLS 16. Almost all of them were also present in simian viruses. It is worth mentioning the following particularities for some CLSs: CLS 2 was detected twice in most HIV-1s, all CPZ and mandrill (pol and nef genes) and most AGM virus genomes (nef gene). It was observed only once (nef gene) for most HIV-2s, Macaque and Sooty Mangabey viruses. Besides, the particular Sykes' monkey virus presented the single CLS 2 in the vif gene. When viral genomes did not possess the second CLS 2 (nef gene), CLS 16 (from which CLS 2 derived) was detected in the pol gene. For CLS 4 (gag gene), a maximum of three transitions (16.7%) and sometimes four transitions (22.2%) were introduced in the computation. In the control viruses, this sequence was only detected in one Herpes virus after three permitted transitions. When reaching four transitions, it were detected in almost all lentiviruses and in some negative genomes. While CLS 3 (pol gene) as well as CLS 4 was found in many genomes in most viral families, they were the only ones present in the four simian D-particle-forming viruses whose presence does not develop AIDS-like syndrome. CLS 6 (pol gene) was present in all the primate viruses, except the Sykes' monkey's ones.
3.2 Sequences common to a maximum of HIV-1 and SIV isolates
These sequences corresponded to CLS 7 up to CLS 15, in addition to those common to a maximum of HIV-1s and HIV-2s. The pattern of detection of HIV-1s was also found for CPZs except that CLS 13 and 14 were missing. CLS 7, 8 and 9 were only detected in HIV-1s and in CPZ viruses. In particular, CLS 10 was detected in most HIV-1s (pol gene), sometimes twice for 34/141 positive strains (gag and pol genes). It was not found in 5/6 HIV-1-O viruses (AF407418, HIM302646, HIM302647, HIVANT70C and HIVMVP5180) but was in AF407419. When CLS 10 was present, its position highly varied in all CPZ viruses (pol gene), the two caprine viruses (vif gene), HIV-2s and Sooty Mangabey (env gene), and Mandrill viruses (pol gene). CLS 13 was present in most HIV-1 genomes (env gene), except for 12 out of 155 HIV-1s including the six HIV-1-O viruses, and was found twice in 40 out of 155 ones (env and pol gene). CLS 14 only found in most HIV-1s was located mainly in gag gene and gradually detected (66% to 85%) when the admitted transitions varied from 1 to 7. CLS 15 (pol gene) was specific to most HIV-1s, but was absent from all the HIV-1-O group, and was present in one CPZ virus.
3.3 Sequences common to a maximum of HIV-2 and SIV isolates
These sequences correspond to CLS 17 up to 21, in addition to those common to a maximum of HIV-1s and HIV-2s. The five CLSs characteristic of HIV-2s were also conserved in most simian viruses except for CPZ, Mandrill and Sykes' monkey viruses. CLS 17 was present in most HIV-2s and Macaque viruses (env gene) and sometimes in sooty mangabey and AGM viruses (rev gene). CLS 18 was found in gag gene in all HIV-2, AGM and macaque genomes and in most sooty mangabey and mandrill ones. CLS 19 (gag gene) and 21 (vpx gene) were detected in most HIV-2s and CLS 20 (env gene) in all of them. CLS 19, 20 and 21 were also present in all macaque and sooty mangabey viruses in the gag, env and vpx genes, respectively.
3.4 Sequences found in only SIVs or non-primate lentiviruses
In addition to the sequences common to a maximum of HIV-2s, CLS 22 was characteristic of all AGM and Sykes' monkey lentiviruses in the env and tat overlapping genes, respectively. When present in one of the two Mandrill viruses (SIVMNDGB1), CLS 22 was located at an unusual position in the gag gene.
Non-primate lentiviruses showed less CLSs than primate ones, with 13/28 not present at all (Table 3), bovine lentiviruses showing the minimal number of two CLSs. The missing CLSs concerned particularly those detected in HIV-1s. It is worth mentioning that CLS 2 was uniquely observed in all equine viruses where it was detected once at the unusual position in gag gene. CLS 10 was only present in all caprine viruses (unusually in vif gene) as well as CLS 16 in most feline ones (pol gene). MMy 1® was restricted to all equine and feline viruses.
3.5 Sequences found in other retroviral genomes and negative controls
A few CLSs were displayed in some negative control viral genomes, while the random-generated ten-kilobase-DNA-like structures did not present any of them. CLS 3 was found in 8/9 HTLV-1s together with CLS 4 in 5/9 of them while HTLV-2s did not present any sequence. CLS 3 and 4 were the only ones detected in D-particle-forming simian viruses in the pol and gag genes, respectively. MMy 1® was in 1/6 murine retroviral genomes. One should note that all these different oncornaviruses belong to families that are genetically stable and do not induce immunodeficiency. CLS 1 and 22 were in retroviral spumaviruses (3/5 and 2/5, respectively). CLS 3, 4, 10, 17, 20 and 21 were found in 1/4 herpes viruses. Remarkably, HIV-1s present a cross-transactivation activity on herpes viruses [40,41] as well as HTLV-1s [42]. CLS 18 was in 1/10 picornaviruses while CLS 16 was in 5/13 parvoviruses.
3.6 CLS location on viral genomes
The observed sequences were mapped on all the genomes of the different viral families. From the particular organization of the HIV-1 and HIV-2 (Fig. 1), AGM and macaque (Fig. 2), CPZ, feline, equine and D-particle-forming viruses (Fig. 3) and that of sooty mangabey, mandrill and other non-primate lentiviruses (supplementary data), it appears that a given CLS occupied on the viral genome a specific position that was roughly conserved in the different viral families. At first sight, the CLSs were detected mainly in the gag and pol structural genes and, to a lesser extent, in the LTRs, the env structural gene and the nef, vpr, vpu, vpx, vif, rev and tat regulatory genes in a decreasing order.
When analyzing the data among families, the HIV-1 genome displayed the highest number of CLSs that were mainly found in the first half of the genome, covering 5′ LTR, gag and pol genes and a part of vif gene. Particularly, the sequence detection evidenced the p17 and p24 proteins for gag gene and the p31 and p51 proteins for pol gene, while CLS 11 and 13 were found in the gp120 protein of env gene (see Tables 2 and 3). Moreover, it was noticeable that CLS 7 was at the hinge of the two LTRs. In HIV-2 genome, CLSs were also mainly related to gag, pol and env genes. HIV-2 and SIV genomes presented similar organizations, but some differences led us to differentiate the SIVs in several categories (Figs. 2 and 3; Supplementary data). For example, CPZ viruses exhibited a striking analogy with HIV-1s, while AGM, macaque and sooty mangabey genomes showed a CLS organization rather similar to that found in HIV-2s, confirming molecular data. CPZ viruses also presented such a similarity with HIV-1s at the molecular level, yet they belonged to the simian viruses concerning the immunological characterization (e.g., [1,8,43]). Besides, the Sykes' monkey virus appeared to be particular since it presented only six CLSs, four of them being at the hinge of the pol/vif genes. The D-particle-forming viruses showed the simplest organization with only CLS 3 and 4 that framed the beginning of pol gene. For the non-primate mammal viruses, the data must be cautiously interpreted, because the low number of studied genomes was not representative for some of these families. However, it is noteworthy that their detected sequences were mainly situated in the gag-pol region. The CLSs of these viruses whose number increased from bovine, equine, ovine/caprine, ovine, feline and finally up to caprine genomes showed a similar pattern (Fig. 3; Supplementary data).
4 Discussion
4.1 Specificity of CLSs and recombinations
Lentiviral genomes contained 28 CLSs allowing them to divide into regions corresponding to ‘evolutive cassettes’ that defined viral specific subtypes. About one third of CLSs were common to almost all primate lentiviral genomes. As to CLSs specific to HIVs, their detection fitted with the known immunological families. Maps of the viruses that can be reconstructed from building blocks delimited by CLSs were specific to each viral type, though they presented a similar high density in the gag-pol region. The revealed homology in gene location of CLSs between HIVs and simian viruses confirmed the separation into two categories (HIV-1/CPZ viruses, HIV-2s/other SIVs). A clear barrier between primate and other mammal viruses appeared, due to shifting locations for some CLSs in the non-primate ones.
Many studies describe lentiviruses as mosaic viruses and numerous examples of recombination between different HIV-1s strains have been reported [6,7,13–15,19–34]. The correlation between wide recombination segments (whose list is not exhaustive) and CLSs acutely localized in the same regions is shown in Table 4. Most of CLSs located in HIV-1s (82%) corresponded to such reported sequences, an underestimated value since multiple recombinogenic segments lacking of precise location (e.g., [35,36]) are not mentioned. CLS 2 and 4 as well as MMy 28 were situated in the HIV-1 recombination sequence shown in the gag-pol region [21]. Other domains contained CLS 2 and CLS 7 in the nef-LTR region [19], MMy 1 (LTR), MMy 3 (gag gene) and MMy 31, as well as CLS 9 (pol gene) in a study using HIV-1 strains deleted in the env gene [22]. In complete HIV-1 genomes, recombinogenic segments have been reported in the pol gene at RT level (p51 protein), to which corresponded CLS 8, 9, 10 and 12, and in the env/nef common part, containing CLS 2 [20]. They concerned also the gag (p17 protein), env (gp 41 and 120 proteins) and nef genes that carried MMy 3 as well as CLS 10 and 14, CLS 11 and 13, and CLS 2 and 7, respectively [6]. Recombinogenic segments corresponded to CLSs 11 and 13 in env gene (gp120 protein) and CLS 14 in the tat/rev overlapping region [34]. Among the 22 CLSs present in HIV-1s, 82% belonged to the large recombination domains cited above. MMy 4′ and CLS 5 that corresponded to gag p24 protein (core protein) and CLSs 6 and 15 that corresponded to pol p31 protein (integrase) have not been correlated until now to recombinogenic segments.
Correlation between recombinogenic segments described in HIV-1s and CLSs
| Reference number | Gene location of reported recombinogenic segments | CLSs detected in corresponding segments |
| [22,25] | LTR | MMy 1 |
| [6,15,21,22,26–28] | gag | MMy 3, 28, CLS 4, 10, 14 |
| [6,7,14,20,22–24,29–31] | pol | MMy 31, 32, CLS 1–3, 8–10, 12, 16 |
| [13,26,27,30,32] | vpr/tat/rev | CLS 14, 17 |
| [6,20,26,27,33,34] | env | CLS 11, 13 |
| [7,19] | env/nef | CLS 2, 7 |
| [19] | nef/ LTR | CLS 7 |
CLSs were characterized by their nucleotide composition and exhibited at first look a clear gap in detection between primate and other mammal viruses. In fact, well-conserved CLSs can represent a signal specific of the strain, the sub-group, the group or the family of virus to which they belong. These sequences can be classified as a function of the possible recombination events they could induce: ‘HIV-1 type’ between the different HIV-1 and CPZ strains; ‘HIV-2 type’ between the different HIV-2s, macaque and sooty mangabey viruses and the feline ones; ‘simian type’ between AGM, mandrill and Sykes' monkey viruses; ‘lentiviral type’ between approximately all mammal lentiviruses; ‘retroviral type’ between almost all retroviruses. Thus CLSs can be considered as the ‘identifying self’ of the viruses and their presence permitted the denomination of ‘viral self’, which could be a ‘species self’ (an HIV-1-type, for example), or an ‘inter-species self’ – ‘lentiviral’ or ‘retroviral’ self.
4.2 Genome flexibility
Our global study of the entire lentiviral genomes suggested the involvement of recombinations in genome flexibility. It allowed us to postulate that if one recombination induced the formation of one variant, the association of series of related variants formed one subtype. The latter was produced by recombinations between distinct variants that could cause or not the emergence of a new subtype. The genomic flexibility is associated with the viral-derived DNA sequences that can recombine [1,19,30] and/or produce complementing and/or recombining RNAs [4,44,45]. Once several elements of the viral genome have penetrated into a cell – and sometimes together with HIV DNA pieces carried by virions [46] – they may be rearranged to possibly elicit productive infection in a new target tissue. For example, CLS 11 and CLS 13 (HIV-1 env gene) were located at the level of the V3 and V4 variable loops of the gp120 cell-binding protein, respectively corresponding to the recombinogenic segments previously described [6,20,26,27,33,34]. This CLS 11/CLS 13 tandem seems to correspond to an important building block for the creation of a new cellular tropism. The V3 domain is critical for chemokine-mediated blockade of infection [47] and the V3 and V4 regions that are separated by the C3 constant domain represent the targets of many trials for the establishment of a candidate vaccine.
A productive viral dissemination has been revealed that evidences recombinations implying independent gene evolutions [1,14,30], and possibly leads to a new gene acquisition [48,49]. Such genomic divergences both maintain viral specificity and allow the emergence of new families raising the question: are these divergences selected by the specificity or are they specificity-selectors while maintaining the viral integrity? So a new viral cell tropism can be created that correlates with the use of the CCR5 or CXCR4 classical co-receptors [50,51] or a postulated new one [52].
4.3 Correlation between some CLSs and known functions
Another essential step was to link the determined landmarks of genomic flexibility with precise viral functions. It is worth mentioning that CLSs 16 and 2 have a very close function. CLS 16 was a part of the cPPT involved in the initiation of lentivirus reverse transcription [53] where DNA synthesis enhances DNA/DNA recombination [54]. CLS 2 represented the well-conserved part of the distal PPT, and perhaps the action site, while the neighbouring sequences which varied highly from a virus to another one might constitute a specific recognition site for the reverse transcriptase associated with a given virus. HIV-1s displayed two CLS 2 at positions shown to be the cPPT (pol gene) [44] and the distal PPT (nef gene) [53]. Also, HIV-2s revealed a slightly different organization, since CLS 16 was part of the cPPT (pol gene) and CLS 2 part of the distal PPT when the nef gene overlaps the 3′ LTR. The role in recombination of CLSs situated in the conserved PPT was emphasized by the determination in the pol gene (protein p31, integrase) of a recombinogenic segment that could affect the viral productive cycle [14].
Another interesting point concerns the sequence structure which could also indicate a specific additional function for some CLSs. For example, the presence of the repeated AATT motif inside CLS 15 (3 motives) and 13 (2 motives) is similar to a folding-like inducer of the DNA [55]. CLS 15 presented the noticeable triplicate AATT structure (AAACTAAAGAATTACAAAAACAAATTACAAAAATT) neighbouring CLS 6 to form a termination structure. In view of the structure of the CTS (pol gene, p31 protein) [56], CLS 15 together with CLS 6 showed the AAAAATT and AAATTTT motives corresponding respectively to 1 and 2, strong and weak, stop signals [57,58]. The CTS approximately represents the succession of these two sequences.
4.4 Possible implication of CLSs in lentiviral ontology and ontogeny
The raison d'être of a CLS – simply recombining, participating in the genetic expression or both – can imply important differences in the detected sequence function and/or viral ontology such as generation of a new subtype (e.g., [26,44]). CLSs specificity revealed that two kinds of recombinations seemed to coexist. One involved sequences mostly common to all the lentiviral genomes separated by large domains, which could allow interspecific recombinations. The other type of recombination involved sequences that within a same family were separated by short distances or even overlapped as shown in HIV-1s for CLS 10 and 14 (gag gene). These findings led us to discriminate between restricted or expanded specificity. In a gene-to-gene study on gag and env genes, wide segments have been described where recombinations could be present, which implied that one recombinant virus characterizes one subtype [12]. The multiplicity of viral subspecies present in the same infected host may be a cause and/or a consequence of the recombination phenomena [1,3,4,30], the generation of recombining strains increasing this process.
The presence of CLSs is not directly associated with the role of a gene since a recombination can be either intra- or intergenic. Such a process involves two adjacent or distant CLSs that can be situated in the same gene or in two different genes. Considering all the possibilities of recombination that happen during an infection, the viability of the newly recombinant genome is the single criterion of selection. The cascade of slow genomic divergences is an integral part of the viral ontogeny to ensure long-term survival. Such genetic approaches could benefit from the defined CLSs, whose characteristic is to be well-conserved and to keep functional the viral genome. Thus the mechanism that maintains the genomic flexibility would be an excellent tool to impede viral growth, particularly when sequences implied in recombination and genetic expression are modified or blocked. As an ad absurdum argument, D-particle-forming viruses that do not cause immunodeficiency presented a single sequence CLS 3 (pol gene), and occasionally CLS 4 (gag gene), a situation leading to the suppression of some necessary specificity steps. These two CLSs were the only sequences detected in HTLV-1s showing a genetic stability like that in HTLV-2s yet without CLS, which suggests that they could play a role in gene exchange between oncorna- and/or retroviruses.
5 Conclusions
During lentiviruses adaptation to changes caused by drug administration [2,45,59,60] or by environmental conditions to ensure continued reproduction, the viral propagation beyond a critical level is constantly in a situation of flux. Thus, CLSs can allow the spontaneous replacement of defective variants with newly ‘recruited’ recombined (or complementing) HIV genomes. The high degree of divergence is a vital part of the viral ontogeny, and recombinations induce sustained viral multiplication allowing the environment to select the most efficient genomic alternative. This emphasizes the importance of the number, the position and the specificity of CLSs on the viral genome, especially since most of them fitted with recombinogenic segments already described. The biological role validity of some CLSs not associated until now with a known function had to be checked in vitro, for example by reverse genetic experiments that could reveal the importance of the different sequences. CLSs, which represent essential landmarks of genomic flexibility, may become key targets for the establishment of new drug and/or gene therapies that can escape the resistances encountered with treatments presently in use.
Acknowledgements
We thank Dr Christiane Plas for critical reading of this manuscript and for helpful discussions.
Appendix A
The studied genomes obtained from the Genbank reference database were as follows. The numbers in parenthesis and in italic represent the number of genomes studied in each viral family.
A.1 Immunodeficiency lentiviral genomes
Human viruses (171): HIV-1 (155): AB023804, AB032740, AB032741, AB049811, AB052867, AB052995, AF004394, AF004885, AF033819, AF042100, AF042101, AF042106, AF049494, AF049495, AF061640, AF061641, AF061642, AF064699, AF067154, AF067155, AF067156, AF067157, AF067158, AF067159, AF069140, AF070521, AF075719, AF084936, AF086817, AF107770, AF107771, AF110959, AF110960, AF110961, AF110962, AF110963, AF110964, AF110965, AF110966, AF110967, AF110968, AF110969, AF110970, AF110971, AF110972, AF110973, AF110974, AF110975, AF110976, AF110977, AF110978, AF110979, AF110980, AF110981, AF119819, AF119820, AF133821, AF164485, AF192135, AF193277, AF197338, AF197339, AF197340, AF197341, AF224507, AF256205, AF256206, AF256207, AF256211, AF259954, AF259955, AF286236, AF286365, AF289548, AF289549, AF289550, AF290027, AF290028, AF290029, AF290030, AF321523, AF332867, AF385934, AF385935, AF385936, AF407418, AF407419, AF408626, AF408627, AF408628, AF408629, AF408630, AF408631, AF408632, A04321, A07867, HIM237565, HIM245481, HIM271445, HIM291719, HIM302646, HIM302647, HIVANT70C, HIVBCSG3X, HIVBRUCG, HIVCAM1, HIVELICG, HIVF12CG, HIVHXB2CG, HIVIBNG, HIVJRCSF, HIVMALCG, HIVMNCG, HIVMVP5180, HIVNDK, HIVNL43, HIVNY5CG, HIVOYI, HIVPV22, HIVP896C, HIVRF, HIVSF2CG, HIVTH475A, HIVU455A, HIVU43096, HIVU43141, HIVU46016, HIVU51188, HIVU51189, HIVU54771, HIVU69584, HIVU69585, HIVU69586, HIVU69587, HIVU69588, HIVU69589, HIVU69590, HIVU69591, HIVU69592, HIVU69593, HIVU71182, HIVU86780, HIVYU2, HIVYU10, HIVZ2Z6, HIV-1U12055, HIV-1U21135, HIV-1U23487, HIV-1U26942, HIV-1U34603, HIV-1U34604, HIV-2132, HIV6287, NC_001802, U88822.
HIV-2 (16): AF082339, HIVTRENGE, HIV-2BEN, HIV-2CAM2, HIV-2D194, HIV-2ISY, HIV-2MDS, HIV-2NIHZ, HIV-2ROD, HIV-2ST, HIV-2UC1GNM, HIV-2U22047, HIV-2U27200, HIV-2U38293, HIV7312A, NC_001722.
Simian viruses (33): Chimpanzee (3): AF103818, AF115393, SIVCPZGAB. African Green Monkeys (9): NC_001549, SIU04005, SIU58991, SIVAGMAA, SIVAGM3, SIVAGM155, SIVAGM385, SIVAGM677A, SIVREV. Macaque (8): RESIVMXX, SIU72748, SIVMM1A11AA, SIVMM239, SIVMM251, SIVMM32H, SIVMNE, SIVSTM. Mandrill (2): AF328295, SIVMNDGB1. Sooty Mangabey (10): AF077017, AF334679, RESIVSMM, SIVGAGAA, SIVGAGAB, SIVSMMPBJA, SIVSMMPBJB, SIV6P6, SIV6P9, SIV6P12. Sykes' monkey (1): SIVCOMGNM.
Non-primate mammal viruses (33): Bovine (2): BIM127, NC_001413. Caprine (2): AF322109, CEAVCG. Equine (11): AF016316, AF028231, AF028232, AF033820, AF247394, AF327877, AF327878, EIAVCG, M87581, NC_001450, U01866. Feline (9): AF474246, FIU11820, FIU56928, FIVCG, FIVGVEPX, FIVPPR, FIVZ1, NC_001482, PLU03982. Ovine (3): AF479638, NC_001511, OLVSAOMVCG. Ovine/Caprine (6): NC_001452, VLVCG, VLVCGA, VLVGAGA, VLVLV1A, VLVLV1B.
A.2 Other retroviral genomes
Spumaviruses (5): FSU85043, HFVHFVCG, HSU21247, SFU04327, SFVGENOME.
Oncornaviruses (25): HTLV-1 (9): AF033817, AF042071, AF139170, AF259264, HL1PROP, HL1RHK30X, HTU19949, HTVPRCAR, NC_001436. HTLV-2 (9): AF074965, AF139382, AF326583, AF326584, AF412314, HL2G12GNOM, HL2V2CG, HTLVCGE, NC_001488. Murine viruses (7): HUMER41, MLFCG, MLFRO, MLMCG, MLM124, MLOCG, MMTPROCG.
D-particle-forming viruses (4): AF033815, NC_001550, SIVMPCG, SIVRV1CG.
A.3 Negative control viruses
Herpes viruses (4): HE1CG, HHV6AGNM, HSECOMGEN, IH1CG.
Other viruses (65): Human and Animal viruses (47): Adenovirus (1): ADRCG. Coronaviruses (6): CCOINSAVC, IBASPMNCF, IBVCD61, MHVDIPORF, PTGPRCVSF, TGVTFIVIR. Filoviruses (2): EBORNA, MVREPCYC. Flaviviruses (12): DENT1SEQ, DVU88535, DVU88536, DVU88537, JEU47032, JEVBEICG, JEVSAA, WNFCG, YFU17066, YFU21055, YFU21056, YFU54798. Parvoviruses (13): AA2CG, GPU25749, HOU34255, MDPCGA, MOU34253, MOU34254, MPU12469, MVMPCG, MVU34256, PARH1, PPU44978, PVCY1A, PVDCG. Picornaviruses (10): ETU22521, ETU22522, HPAACG, HRV, PIP03XX, POL1, POL2CG1, POL2LAN, POL3L12CG, SVDG. Rhabdoviruses (3): IHNCG, SYECG, VSVCG.
Vegetal viruses (18): Geminiviruses (5): MBGCG1Z, MBGCG2Z, MTGA, MTGB, MZS Potyviruses (2): TEVCGHAT, TEVGEN. Tobamoviruses (5): BRU03387, MTVCG, MTVVCG, TMGCG, TMVCG. Tombusviruses (1): TBSACG. Tymoviruses (3): MTYCLCGA, TYMVCG, TYMVXX. Necroviruses (2): TNSCG, TNSDNSC.
Appendix B Supplementary data
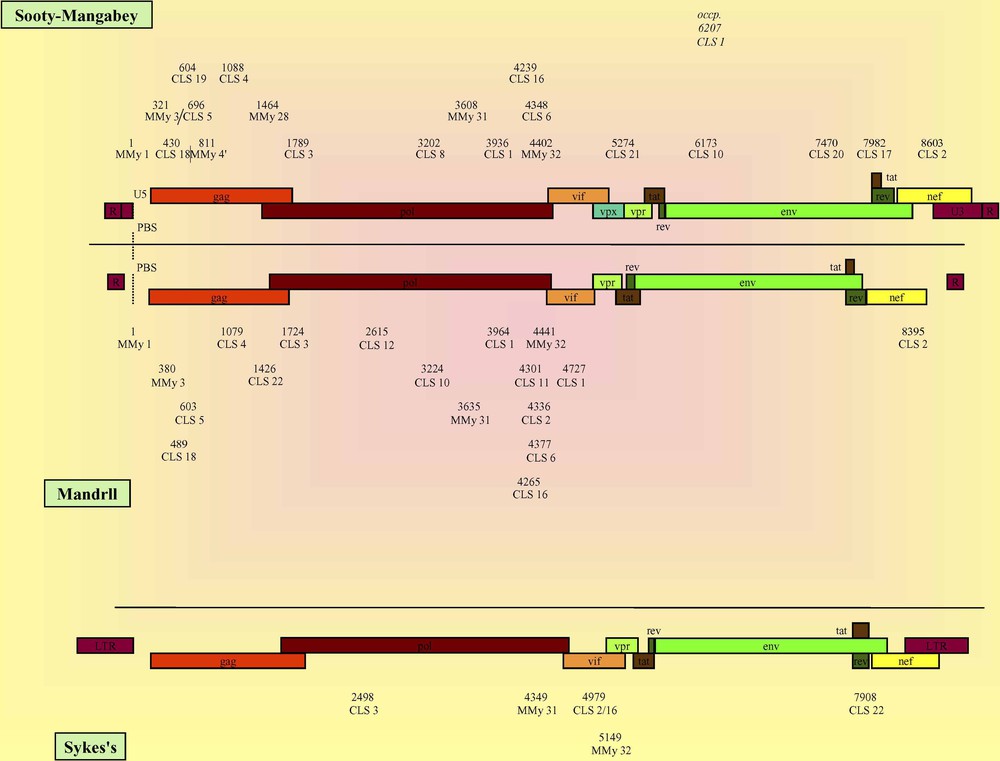
CLSs localization and genomic organization of Sooty Mangabey, Mandrill and Syke's viruses. The numbers represent the relative positions of the detected CLS calculated versus that of the MMy 1 except for Syke's viruses which lack MMy 1 whose numbers correspond to the crude positions of the CLS directly referenced from the genomes (see Methods). The reference organization of Sooty Mangabey, Mandrill and Syke's viruses was represented using AF334679, SIVMNDGB1 and SIVCOMGNM viruses, respectively. Occp.: CLS occasionally found at an occasional position. CLSs localization and genomic organization of caprine, ovine, caprine/ovine and bovine viruses. Because all genomes lack MMy 1, the numbers correspond to the crude positions of the CLS directly referenced from the genomes (see Methods). The reference organization of caprine, ovine, caprine/ovine and bovine viruses was represented using AF322109, NC_001511, VLVGAGA and NC_001413 viruses, respectively.