

1 Introduction
Les maladies cardiovasculaires constituent l'une des principales causes de morbidité et de décès dans les pays industrialisés, et l'âge est l'un des principaux facteurs de risque de leur développement. Le vieillissement de l'organisme est associé à des modifications à la fois structurelles et fonctionnelles de l'arbre vasculaire. En dehors des altérations qui conduisent à la formation et à l'évolution de lésions athéromateuses, l'artériosclérose liée au vieillissement se caractérise par une rigidification et un épaississement de la paroi artérielle et peut avoir un retentissement au niveau cardiaque. Les processus biologiques mis en jeu font appel schématiquement à trois grandes phases : le dysfonctionnement de l'endothélium, le remodelage de la matrice extracellulaire pariétale et la calcification de la média. Bien que ces processus soient interdépendants de ceux qui conduisent à l'apparition de lésions athéromateuses, l'artériosclérose liée à l'âge possède des caractéristiques qui lui sont propres. L'objectif de cette revue est de faire le point sur la physiopathologie du vieillissement artériel et les mécanismes biologiques qui l'accompagnent en précisant ce qui distingue les lésions dues à l'âge de celles qui sont observées au cours du processus athéromateux.
2 Histophysiologie de l'arbre artériel
La paroi artérielle est classiquement décrite comme la succession de trois couches : l'adventice, la media et l'intima. Au sein de ces différentes couches, il existe de nombreuses interactions entre les cellules (pour une revue, voir [1]) et la matrice extracellulaire. Grossièrement, on retrouve principalement des cellules endothéliales au niveau de l'intima, des cellules musculaires lisses dans la média et des péricytes et des fibroblastes dans l'adventice. Les autres types cellulaires que l'on peut retrouver dans les vaisseaux sont moins bien caractérisés [2]. La présence de macrophages est quant à elle plutôt liée à des situations inflammatoires et pathologiques.
Au niveau de l'intima, la monocouche de cellules endothéliales forme l'endothélium qui a un rôle actif central dans l'interface sang vaisseau (cf. Section 6.1). Dans la média, on retrouve essentiellement des cellules musculaires lisses des vaisseaux (CMLV) dont la principale fonction est le maintien du tonus et la résistance vasculaires. L'adhésion des CMLV contractiles à la matrice extra cellulaire (MEC) est renforcée par de nombreuses protéines pour supporter les tensions considérables et les forces hémodynamiques qui s'exercent sur la paroi. En ce qui concerne l'adventice, les principales cellules qui la composent sont les fibroblastes.
Toutefois cette répartition cellulaire par couches n'est pas figée et au cours de certaines pathologies, on observe des migrations cellulaires et des changements phénotypiques. Les CMLV ont par exemple un rôle important dans la réponse au stress et dans la réparation de la paroi au cours des lésions vasculaires. Cela nécessite une dédifférenciation des CMLV qui changent de phénotype pour devenir mobiles et acquérir une activité de synthèse particulière. Cette nouvelle mobilité nécessite une modification des interactions avec les molécules d'adhésion pour permettre aux CMLV de se désolidariser de la MEC [3]. De même, les fibroblastes de l'adventice joueraient un rôle précoce dans le remodelage artériel. Même si les données sont encore parcellaires car les recherches dans le domaine se focalisent surtout sur les modifications intima-média, ils pourraient, à la suite de lésions vasculaires, proliférer et migrer après s'être différenciés en myofibroblastes. Les cellules de l'adventice seraient également une source importante de radicaux libres agissant sur les cellules musculaires lisses. Cependant, bien que tous les types cellulaires présents dans l'artère (cellules endothéliales, musculaires lisses et de l'adventice) produisent également des radicaux libres oxygénés (RLO) en quantité variable en réponse à divers stimuli, il est admis que la plupart des RLO dans la paroi des vaisseaux provient des macrophages [4]. Source importante de MMPs (matrix metalloproteinases), ces derniers participent également au métabolisme de la matrice extracellulaire (cf. Section 6.2). D'autre part, les macrophages interviennent au cours de la maladie athérothrombotique dans toutes les étapes de la pathologie (initiation, pérennisation des lésions, complication de la plaque). Les monocytes-macrophages, après avoir pénétré dans l'espace sous-endothélial, vont se différencier en macrophages matures puis en cellules spumeuses qui sont capables d'internaliser des lipides et des protéines anormalement modifiées. Les cellules spumeuses peuvent provenir également des CMLV qui ont migré de la média vers l'intima [5].
Le système artériel est schématiquement composé d'artères élastiques de gros et moyen calibres et d'artères musculaires périphériques de petit calibre. Les artères de gros et moyen calibres, centrales dans l'arbre artériel, ont une intima développée, caractérisée par une richesse de la MEC en élastine et en collagène, alors que les artères de petit calibre, distales, ont une couche de cellules musculaires lisses plus développée. Les fibres élastiques ont un rôle majeur dans la résistance mécanique des vaisseaux aux pressions faibles, alors que les fibres de collagène contribuent à la résistance artérielle aux pressions élevées [6].
Les grosses artères élastiques participent à la bonne distribution sanguine dans l'ensemble de l'organisme en amplifiant la pression de sortie du ventricule. En effet, la pression systolique est plus importante dans les grosses artères des membres supérieurs et inférieurs que dans l'aorte ascendante permettant ainsi de réduire le travail cardiaque. Les vaisseaux de plus petit calibre tamponnent le flux artériel et les variations de pression induites par la contraction intermittente du ventricule gauche, et transforment les variations pulsatiles du flux en un flux continu pour les artères périphériques, permettant ainsi l'oxygénation des tissus périphériques. La perte de l'élasticité est responsable d'une rigidification artérielle centrale, qui modifie la pression artérielle sanguine : celle-ci est essentiellement déterminée par les résistances périphériques vasculaires chez le sujet jeune et par la rigidité artérielle chez le sujet âgé [7,8].
Dans le système artériel, le fonctionnement rythmique de la pompe cardiaque est à l'origine d'une onde de pouls (Fig. 1) qui est déterminée par deux composantes : l'une dépend de la contraction ventriculaire et l'autre, moins importante, d'ondes de réflexion remontant l'arbre artériel, en provenance de points où la résistance à l'écoulement change brusquement, au niveau des bifurcations vasculaires notamment. Lors d'une perte d'élasticité de la paroi artérielle, la vitesse de propagation de l'onde de pouls vers la périphérie s'accélère, de même que celle des ondes de réflexion. Ainsi, le retour prématuré des ondes de réflexion dans l'aorte peut survenir à la fin de la systole, entraînant une augmentation de la pression systolique centrale [7,8]. La pression diastolique est également influencée par la distensibilité pariétale artérielle. A une fréquence cardiaque donnée, la pression régnant dans l'artère diminue en effet d'autant plus rapidement et d'autant plus bas que la paroi du vaisseau est plus rigide. C'est pourquoi l'âge et le vieillissement artériel sont responsables à la fois d'une augmentation de la pression systolique et d'une diminution de la pression diastolique [9,10] (Figs. 1 et 2).
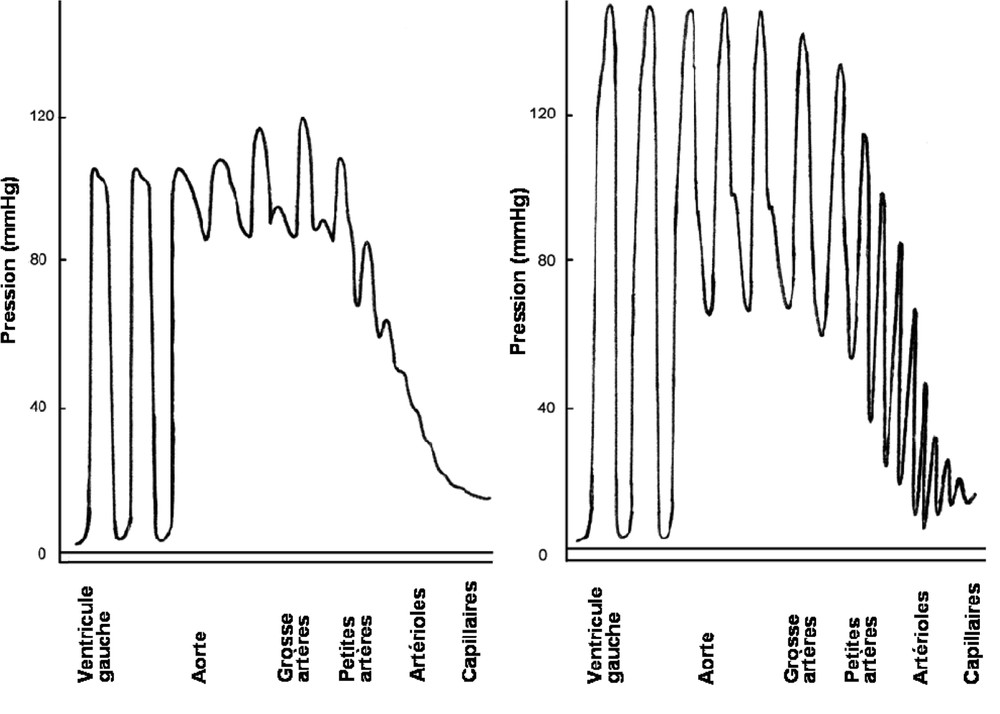
Changement de pression pulsatile dans l'arbre vasculaire. Représentation schématique de la pression pulsatile entre le ventricule gauche et les capillaires chez un sujet jeune (à gauche) et chez sujet plus âgé (à droite) avec une rigidité artérielle. Chez le sujet âgé, les pulsations ne sont pas absorbées dans les grosses artères et donc sont présentes au niveau de la microcirculation (d'après [8]).

Effet de l'âge sur la pression sanguine systolique et diastolique. Les pressions sanguines diastolique et systolique augmentent en parallèle jusqu'à l'apparition de changements physiopathologiques. Après 50 ou 60 ans, la pression systolique continue d'augmenter alors que la pression diastolique commence à diminuer (d'après [10]).
3 Les processus d'athérosclérose et d'artériosclérose
Ces deux processus d'évolution structurale et fonctionnelle de l'arbre vasculaire sont à la fois physiologiques car ils interviennent inexorablement au cours de l'avancement de la vie, et pathologiques dans la mesure où ils peuvent être responsables d'évènements cliniques (cardiaques, cérébraux, périphériques) porteurs de morbidité et de mortalité. Ils sont également interdépendants et très intimement liés, et certains des évènements cellulaires et moléculaires qui les caractérisent sont communs. C'est pourquoi le terme d'artério-athérosclérose peut être utilisé pour définir l'ensemble des processus délétères qui déterminent les modifications de l'arbre artériel au cours du vieillissement.
3.1 L'athérosclérose
L'athérosclérose est définie par l'Organisation mondiale de la santé (OMS) comme « une association variable de remaniements de l'intima des artères de gros et de moyen calibre consistant en une accumulation focale de lipides, de glucides complexes, de sang et de produits sanguins, de tissu fibreux et de dépôts calcaires ». Ce remodelage de l'intima s'accompagne de modifications de la média [11]. Le processus athéromateux débute dès l'enfance, évolue de façon infraclinique en engendrant progressivement des lésions et peut aboutir, beaucoup plus tard dans la vie, à une sténose artérielle. A la différence du processus artérioscléreux (cf. infra), l'athérosclérose est formée de lésions artérielles localisées, qui s'accompagnent d'une diminution du calibre artériel et qui siègent principalement au niveau des artères élastiques.
La description anatomo-pathologique actuelle de l'athérosclérose retient trois stades évolutifs : la strie lipidique, la lésion fibrolipidique et la lésion compliquée. Une classification beaucoup plus détaillée a été proposée par Stary et retenue par l'AHA [12]. Plusieurs événements physiopathologiques du processus athéroscléreux, successifs ou concomitants, peuvent être distingués [13] :
- – la rétention de lipoprotéines athérogènes dans l'intima, après leur passage transendothélial et leur fixation à des constituants de la matrice extracellulaire intimale ; l'activation de l'endothélium artériel et le dysfonctionnement cellulaire qui en résulte contribuent à ce processus ;
- – la modification physicochimique des lipoprotéines retenues, sous l'influence d'un stress oxydant local exacerbé ;
- – le recrutement des monocytes circulants, sous l'influence de facteurs chimiotactiques et grâce à l'expression accrue de molécules d'adhésions endothéliales, et la transformation ultérieure des cellules recrutées en macrophages puis en cellules spumeuses ;
- – la formation de la chape fibromusculaire constituée principalement de cellules musculaires qui ont migré à partir de la média et ont proliféré dans l'intima, imposant un remaniement de la matrice extracellulaire intimale en faveur de sa dégradation ;
- – la calcification de la plaque, par constitution et dépôt de composés calciques au niveau intimal et médial, rendant celle-ci friable et apte à la rupture sous l'influence de facteurs hémodynamiques mécaniques.
Les évènements thrombotiques locaux, liés à la rupture des lésions et au déversement de composants matriciels prothrombotiques dans le lit artériel, sont infracliniques dans 90% des cas. Comme cela a été bien démontré au cours des dix dernières années, un déterminant fondamental de la progression athéroscléreuse est l'instauration d'un état pro-inflammatoire de faible intensité mais permanent et chronique, qui entretient les dysfonctionnements cellulaires et moléculaires locaux au sein des lésions athéromateuses et favorise ainsi leur évolution et leur fragilisation. L'aboutissement critique de ces lésions athéromateuses est une sténose artérielle et/ou la rupture de la plaque lipidique calcifiée qui vont conduire à une ischémie des territoires situés en aval de la lésion.
3.2 L'artériosclérose
L'artériosclérose est définie comme « l'épaississement et la perte d'élasticité de la paroi des artères de toutes tailles ». A l'inverse de l'athérosclérose, la physiopathologie de l'artériosclérose a été moins étudiée. Même s'il existe de nombreux points communs entre les deux pathologies (inflammation, calcification, augmentation de l'épaisseur intima-média (EIM) liée au remodelage matriciel extracellulaire...), on peut distinguer une artériosclérose non athéromateuse dont les caractéristiques physiopathologiques sont différentes de l'athérosclérose proprement dite (Tableau 1).
Principales différences entre l'athérosclérose et l'artériosclérose. Le tableau rapporte de façon résumée et non exhaustive les grandes caractéristiques différentielles des deux pathologies.
L'artériosclérose est le résultat de modifications non pas focales en certains points de l'arbre vasculaire, mais généralisées sur l'ensemble de la longueur du système artériel. Elles se traduisent par un épaississement et une perte d'élasticité liée à des modifications structurales de l'intima (migration des cellules musculaires lisses de la média, remaniement de la matrice extracellulaire) et de la média (hypertrophie des cellules musculaires lisses, accumulation des collagènes, de la fibronectine et formation de dépôts calciques). Il en résulte une diminution de la compliance des artères élastiques et une modification de la fonction endothéliale [14]. L'artériosclérose ne conduit pas à une diminution de la lumière des artères de gros et moyen calibres comme dans l'athérosclérose mais plutôt à une augmentation du diamètre de ces artères [8]. Comme nous l'avons vu, les artères ont un rôle important dans l'amortissement de la pression sanguine à la sortie du ventricule et participent à l'efficacité du système cardiovasculaire [15]. Les troubles de l'amortissement par rigidification de la paroi artérielle que l'on observe au cours de l'artériosclérose vont plutôt avoir des répercussions en amont c'est-à-dire sur la fonction cardiaque [16].
4 Expression clinique de l'artériosclérose
L'aboutissement de l'artériosclérose est la calcification du vaisseau qui conduit à sa rigidification. A ce titre, la sclérose de Mönckeberg peut être considéré comme un exemple caricatural de calcification artérielle chez l'homme. Initialement décrite en 1903 par Mönckeberg, cette pathologie calcifiante de l'arbre artériel est directement liée à l'âge. Au niveau histologique, la présence de dépôts calciques au niveau de la média, et de la lame élastique interne est constante et massive [17].
L'âge et le sexe sont les deux plus importants facteurs de risque de calcification coronaire ; la prévalence de ces calcifications est de 93 à 100% chez l'homme de plus de 70 ans et de 77 à 100% chez la femme de plus de 70 ans [18]. Les conséquences phénotypiques de la calcification vasculaire sont la dilatation et l'augmentation de rigidité des artères élastiques de conduction (tout au moins proximales), ce qui se traduit cliniquement, notamment, par une augmentation importante de la probabilité de complications cardiovasculaires chez des sujets coronariens et un excès de mortalité à cinq ans. L'augmentation de la rigidité artérielle s'accompagne d'une augmentation de la charge de travail du ventricule gauche au travers de l'augmentation de la pression pulsée (qui correspond à l'écart entre les pressions diastolique et systolique) et des ondes de réflexion.
Il existe en effet un lien étroit entre la rigidité artérielle et l'hypertension. Il est important de bien distinguer deux types d'hypertension : celle qui allie hypertension systolique et diastolique et l'hypertension systolique seule [15]. Dans le premier cas, c'est l'hypertension classique que l'on retrouve essentiellement dans une population de gens jeunes ou d'âge moyen. La pression pulsée est donc peu augmentée. Mécaniquement, plus la pression exercée sur un vaisseau est importante, plus il se dilate, et donc plus il se rigidifie. Avec l'âge, l'augmentation de la rigidité des gros troncs artériels contribue à l'augmentation de la pression pulsée [19] qui se traduit par une hypertension systolique sans hypertension diastolique (Fig. 2). A l'accroissement de la charge de travail du ventricule gauche s'ajoute alors une baisse de la perfusion coronaire liée à la diminution de la pression diastolique qui va favoriser encore plus l'hypoxie voire l'ischémie myocardique clinique.
Les mécanismes moléculaires précis de ces remaniements histologiques artériels restent cependant mal connus : une stimulation adrénergique, l'hypervitaminose D et un lien avec une neuropathie ont été suggérés. De même, l'association d'une calcification artérielle intense avec des maladies métaboliques (diabète, insuffisance rénale chronique, ostéoporose,...) a été décrite [20].
5 Exploration et évaluation du vieillissement artériel (pour revue, [21])
L'évaluation de l'état fonctionnel artériel est actuellement réalisée par des examens cliniques et une exploration fonctionnelle non biologique visant à apprécier la vitesse de l'onde de pouls, la pression pulsée, l'EIM et l'état de la calcification artérielle.
5.1 La pression pulsée
La pression pulsée correspond à la différence entre le maximum de pression systolique et le minimum de pression diastolique. Elle est considérée comme un index de rigidité artérielle et comme le plus puissant déterminant de l'EIM et de la dilatation [19]. L'augmentation de la pression pulsée est un marqueur de risque de mortalité totale et cardiovasculaire, mais aussi de morbidité chez le sujet hypertendu essentiel [22,23]. Chez l'homme, l'EIM augmente d'un facteur 2 à 3 entre 20 et 90 ans [24].
5.2 L'ultrasonographie (mesure de la vitesse de l'onde de pouls)
Cette technique permet de mesurer la vitesse de l'onde de pouls et donc d'évaluer la calcification artérielle de manière qualitative et semi-quantitative mais elle ne permet pas de distinguer la calcification de l'intima par rapport à la média. La vitesse de l'onde de pouls correspond à la vitesse avec laquelle l'onde de pression se propage le long d'un segment de l'arbre artériel. Plus le vaisseau est rigide, plus l'onde se déplace rapidement. Cette valeur augmente de façon linéaire avec l'âge quand elle est mesurée au niveau brachial, mais suit plutôt une progression polynomiale de deuxième ordre quand elle est mesurée au niveau aortique, avec une très nette augmentation à partir de 50 ans [15]. Il existe donc un décalage selon que les artères étudiées sont des artères élastiques de gros diamètre ou des artères musculaires de plus petit diamètre. Ainsi, si à âge égal, le diamètre interne de l'artère carotide commune ou de l'aorte est augmenté chez un sujet hypertendu par rapport à un sujet normotendu, celui des artères musculaires distales comme les artères fémorale, humérale ou radiale reste inchangé [19]. La vitesse de l'onde de pouls est un marqueur établi de maladie cardiovasculaire en particulier chez le sujet âgé [25,26]. Au niveau aortique, l'évaluation de la rigidité de l'artère permet notamment de préciser le risque de mortalité totale et de mortalité cardiovasculaire [27] ainsi que la survenue d'accidents vasculaires [28]. Peu onéreuse et n'émettant pas de radiation, l'ultrasonographie est souvent utilisée en combinaison avec la radiographie (rayons X) notamment pour du screening et de la prédiction du risque cardiovasculaire.
Cette technique permet également une évaluation de l'EIM qui est bien corrélée avec les données histologiques. Elle est utilisée le plus souvent au niveau carotidien dans le cas de la recherche ou du suivi de la formation des plaques athéromateuses.
5.3 Radiographie
Cette technique permet d'obtenir des informations sur la localisation de la calcification au niveau de la paroi. La calcification de la média se matérialise par des lésions opaques qui sont visibles tout le long de l'artère alors qu'au niveau de l'intima, on observe plutôt des lésions en plaques avec une opacité variable. La présence de lignes de calcification au niveau des artères iliaques, fémorales, digitales et radiales permet de déterminer des scores de 0 (aucun vaisseau touché) à 8 (tous les vaisseaux touchés). Ces radiographies corps entier (bien que principalement qualitatives) permettent une détermination semi-quantitative de la calcification mais ne sont pas capables de détecter des changements subtils de la calcification au cours du temps. C'est la technique recommandée par la National Kidney Foundation pour les patients en insuffisance rénale chronique.
5.4 Tomodensitométrie
Les deux principales techniques sont l'“Electron Beam Computed Tomography” (ou EBCT) et la “Multi Slice Computed Tomography” (ou MSCT). Elles sont considérées comme les méthodes de référence pour mesurer l'étendue de la calcification artérielle et sa progression grâce à des scores. Les problèmes relatifs à leur utilisation sont leur coût élevé et l'exposition à des quantités non négligeables de radiation (surtout pour l'EBCT). Elles ne permettent pas la différenciation entre la calcification située au niveau de l'intima et celle située dans la media.
5.5 L'échocardiographie
Elle est principalement utilisée pour évaluer la calcification des valves et des autres structures cardiaques.
5.6 La tonométrie d'aplanation
Cette méthode non invasive, réalisée au niveau des artères de distribution superficielle avec l'aide d'un capteur externe, permet de déterminer la pression artérielle de manière fiable et reproductible et d'accéder par le calcul à la mesure de l'onde de pouls.
Actuellement, grâce à ces méthodes d'exploration, trois grands phénotypes de vieillissement artériel pourraient être schématiquement définis :
- – le vieillissement physiologique, qui correspond à l'évolution physiologique des propriétés structurales et fonctionnelles de l'arbre vasculaire ;
- – un vieillissement ralenti, qui se distingue par des altérations réduites des fonctions vasculaires chez un sujet par rapport à la population d'âge identique ;
- – un vieillissement accéléré, caractérisé par une évolution plus rapide de l'altération vasculaire chez un sujet donné par rapport à la population d'âge identique.
Cette détermination du phénotype artériel du patient, d'autant plus qu'il est âgé, pourrait être d'une aide précieuse pour la prévention, la surveillance et le traitement des pathologies liées à l'artériosclérose.
6 Biologie et physiopathologie du vieillissement artériel
Des mécanismes physiopathologiques très généraux interviennent, et parfois de manière très importante dans le vieillissement artériel : l'état inflammatoire chronique de la paroi artérielle au cours du processus athéroscléreux a été largement étudié et constitue un acteur important des altérations vasculaires qui se développent en fonction de l'âge. De même, les processus oxydatifs ( « stress oxydant »), liés à une surproduction d'espèces réactives de l'oxygène ou à une diminution des capacités antioxydantes, extracellulaires ou intracellulaires, contribuent au déséquilibre de l'homéostasie redox, elle-même essentielle à certaines fonctions physiologiques cellulaires [13].
Dans le contexte de la mise en place d'une évolution artérioscléreuse de l'arbre vasculaire, trois évènements méritent particulièrement d'être pris en compte car ils pourraient être à l'avenir les sources de biomarqueurs du vieillissement artériel, et des cibles pharmacologiques potentielles : le dysfonctionnement de l'endothélium, le remodelage de la matrice extracellulaire pariétale et la calcification médiale et ses déterminants moléculaires.
6.1 Anomalie de la vasorégulation endothéliale
L'endothélium artériel est situé à l'interface entre le flux sanguin et le tissu vasculaire et ne constitue pas une simple barrière inerte. C'est un tissu dynamique, hétérogène et disséminé sur la totalité de l'arbre vasculaire qui assure des fonctions de synthèse, de métabolisme et de sécrétion de molécules endogènes bioactives. L'endothélium possède donc un rôle clé chez l'individu sain puisqu'il est capable de répondre à des stimuli physiques (comme les forces de cisaillement) ou chimiques en produisant de nombreuses substances qui interviennent dans la régulation du tonus vasculaire, l'adhésion cellulaire, la résistance à la thrombose, la prolifération des cellules musculaires lisses ou encore l'inflammation de la paroi vasculaire. Il apparaît aujourd'hui acquis que le vieillissement et le dysfonctionnement endothélial artériel évoluent parallèlement (pour revue [29]).
Le vieillissement vasculaire se traduit par des modifications structurales et fonctionnelles de l'endothélium comme l'altération de la vasotension/vasorelaxation physiologique et l'acquisition par l'endothélium d'un phénotype inflammatoire. A ce niveau, le monoxyde d'azote (NO) a un rôle déterminant dans la régulation du tonus vasomoteur. En effet, le NO, qui est produit par la NOSynthase de type III des cellules endothéliales, agit de manière paracrine notamment au niveau des cellules musculaires lisses de la média vasculaire et provoque une augmentation du GMP cyclique. L'inhibition de la contraction des cellules musculaires lisses qui en résulte induit ainsi une augmentation du flux sanguin vers les tissus. Au cours du vieillissement, les altérations de l'endothélium entraînent une diminution de cette production de NO et une réactivité augmentée aux vasoconstricteurs comme l'ET-1, ou l'angiotensine II, d'où une augmentation du tonus artériel. Le dysfonctionnement endothélial est observé dans de nombreux états pathologiques tels que l'athérosclérose, l'HTA, l'insuffisance cardiaque et les maladies thrombotiques artérielles [30]. La majorité des facteurs de risque de ces pathologies cardiovasculaires (âge, hyperlipidémie, hypertension, diabète, hyperhomocystéinémie...) interviennent en diminuant la production endothéliale du NO ou en accroissant son élimination.
Outre son action sur la média artérielle, le NO peut également diffuser vers le compartiment sanguin où ses devenirs sont multiples : d'une part le NO peut agir en inhibant l'agrégation plaquettaire, d'autre part, il existe des interactions du NO avec l'hémoglobine qui conduisent à la formation de la S-nitrosohémoglobine (SNOHb) ou de la nitrosylhémoglobine (NOHb), et avec l'albumine pour former des S-nitrosothiols. Tous ces dérivés sont des formes de transport et de stockage temporaire de NO lui assurant une demi-vie très augmentée par rapport au NO libre. Les hématies pourraient jouer un rôle important dans la dégradation du NO par une réaction de co-oxydation avec l'oxyhémoglobine pour former de la methémoglobine et des nitrates [31]. Ces différentes formes dérivées du NO pourraient être autant de biomarqueurs potentiels de sa production endothéliale.
Le processus de biosynthèse du NO est complexe et fait intervenir de nombreux cofacteurs et systèmes de régulation. Parmi eux, la diméthylarginine asymétrique (ADMA) est un composé endogène issu de l'arginine (précurseur endogène du NO) possédant un effet inhibiteur compétitif sur l'activité NOS. L'ADMA est physiologiquement dégradée par la diméthylarginine diméthylaminohydrolase (DDAH), enzyme particulièrement sensible aux altérations du statut redox cellulaire, donc au dysfonctionnement endothélial artériel (Fig. 3). Une augmentation des concentrations plasmatiques de l'ADMA, en partie liée à une diminution de l'activité de la DDAH, est associée à une diminution de la biodisponibilité du NO ; elle est responsable d'une altération de la production vasculaire de NO, du tonus vasculaire et de la résistance systémique des vaisseaux [32]. Les études expérimentales in vivo chez l'animal et les études cliniques ont montré que l'augmentation des concentrations d'ADMA est associée à des facteurs de risque cardiovasculaire (HTA, dyslipidémie, diabète...) et à l'existence d'une atteinte coronarienne [33].
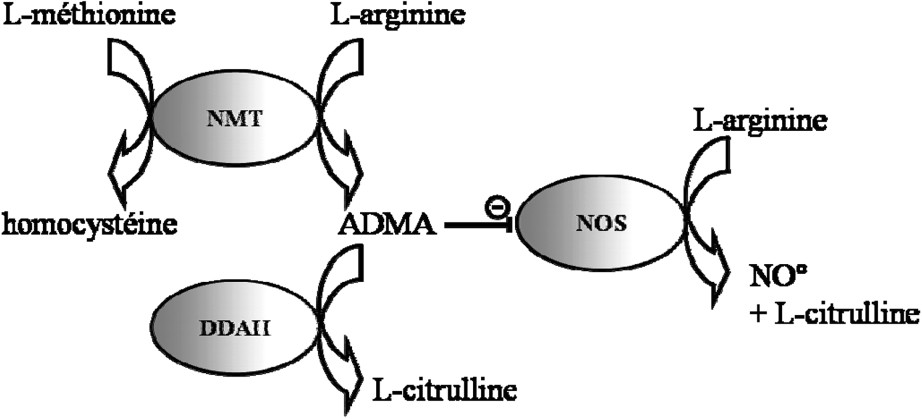
Métabolisme de l'ADMA et conséquence sur la production de NO. La diméthylarginine asymétrique (ADMA), produite par la N-methyltransférase (NMT) à partir de l'arginine, est capable d'inhiber la production de NO par les NOS. L'ADMA est dégradée par la diméthylaminohydrolase (DDAH) en citrulline.
Liée à la synthèse de l'ADMA, l'homocystéine est un acide aminé soufré produit par la déméthylation de la méthionine (Fig. 3). De nombreuses études cliniques ont déjà mis en évidence une corrélation entre une hyperhomocystéinémie et un risque cardiovasculaire augmenté [34]. D'autre part, les patients souffrant d'une hyperhomocystéinémie d'origine génétique ayant une athérosclérose prématurée et une maladie thrombotique veineuse, l'hypothèse d'un effet vasculotoxique direct de l'homocystéine a été proposée [35]. L'homocystéine et l'ADMA partagent donc de nombreux mécanismes physiologiques dont la modification peut favoriser une pathologie vasculaire, via notamment une prolifération des cellules musculaires lisses, une altération de la fonction endothéliale vasculaire NO-dépendante, responsable d'une vasoconstriction et donc d'une HTA.
Pour contrebalancer les effets vasodilatateurs du NO, l'endothéline (ET) a été identifiée, il y a quelques années, comme un puissant vasoconstricteur impliqué dans le tonus vasculaire. On connaît actuellement trois isoformes d'ET, mais c'est l'ET-1 qui prédomine dans le système cardiovasculaire. Ce peptide de 21 acides aminés est essentiellement synthétisé par les cellules endothéliales et agit par voie paracrine sur la contraction des cellules musculaires lisses sous-jacentes. Outre ses effets vasoconstricteurs, l'ET-1 semble également moduler l'expression des NOS et intervenir dans le métabolisme sodique, notamment en cas de déficit du système rénine-angiotensine [36,37].
La régulation du tonus vasculaire est donc un phénomène complexe faisant intervenir de nombreuses molécules qui ont des effets directs ou indirects sur la vasoconstriction ou la dilatation des vaisseaux. NO, ADMA, homocystéine et ET-1 font partie d'un système intégré dans lequel la modification des concentrations d'une ou plusieurs de ces molécules peut entraîner un dysfonctionnement vasculaire. C'est pourquoi l'ensemble des acteurs de la voie du NO sont des candidats intéressants comme biomarqueurs du fonctionnement et par extension, du dysfonctionnement de l'endothélium.
Au delà de ses propriétés vasodilatatrices, le NO peut avoir, selon l'environnement redox, une activité antioxydante ou à l'inverse, prooxydante. En effet, un stress oxydant lié à l'activation chronique de la NAD(P)H oxydase membranaire endothéliale, est à l'origine d'un dysfonctionnement endothélial vasculaire. L'anion superoxyde, formé par l'activation de la NAD(P)H oxydase, peut se condenser avec le NO pour former du peroxynitrite ONOO-, dont les effets sont doubles : inactivation de la manganèse superoxyde dismutase (MnSOD) par nitrosylation des résidus tyrosine de l'enzyme, et découplage de la NOS de la formation de NO vers la formation d'anion superoxyde, par oxydation du cosubstrat de l'enzyme, la tétrahydrobioptérine. De ces deux actions résulte une production d'anion superoxyde accrue et un cercle vicieux privilégiant la formation de peroxynitrite à l'origine d'altérations chimiques de l'ADN [29]. Finalement, une modification de la production du NO peut, par accroissement du stress oxydant endothélial, participer à l'acquisition par l'endothélium d'un phénotype pro-inflammatoire [38]. Ce dernier apparaît sous l'action de stimuli d'origines multiples (produits d'oxydation des macromolécules lipidiques, produits de glycation avancée (AGE)...). Les cellules endothéliales vont exprimer alors des récepteurs médiateurs de l'inflammation (récepteur LOX-1, récepteur RAGE, ...), des molécules d'adhésion facilitant le recrutement monocytaire et donc la désorganisation pariétale, et sécrètent, au même titre que les autres types cellulaires de la paroi artérielle, des cytokines et chimiokines pro-inflammatoires [39,40]. Cette activation pro-inflammatoire de l'endothélium est elle-même à l'origine d'une altération de l'intégrité structurale et fonctionnelle de la MEC sous-endothéliale.
6.2 Le remodelage de la matrice extracellulaire (MEC)
La matrice extracellulaire (MEC) est le composant majoritaire de l'intima et de la média de l'artère normale et constitue le milieu que tous les éléments nutritifs vont traverser, et à travers lequel certaines cellules vont migrer. Elle est synthétisée par les cellules qu'elle entoure et détermine en retour le phénotype de ces cellules. Les interactions cellules-MEC sont très étroites. Quatre grands types de macromolécules constituent la MEC :
- – les fibres de collagène, qui confèrent à la paroi une résistance mécanique aux tissus ;
- – les protéoglycannes, qui ont la capacité de fixer de nombreuses molécules d'eau ;
- – l'élastine, composant majeur des fibres élastiques qui sont organisées dans les artères en lames concentriques parallèles à la surface du vaisseau, et assurant la distensibilité ;
- – les glycoprotéines de structure, comme la fibronectine ou les laminines.
Outre ce rôle de support physique, les protéines de la MEC interagissent avec les différents types cellulaires de la paroi.
Au cours du processus artérioscléreux, le remodelage entraîne des modifications structurales et fonctionnelles de la paroi artérielle et associe au moins quatre processus cellulaires : la croissance, la migration cellulaire, la production de MEC ou sa dégradation, l'apoptose. Ces processus dépendent d'interactions dynamiques entre des facteurs de croissance, des facteurs de l'inflammation produits localement, des molécules vasoactives, et des stimuli hémodynamiques. L'inflammation chronique de la paroi artérielle qui accompagne le vieillissement vasculaire est caractérisée par une hyperactivité des différents types cellulaires présents au niveau de la lésion vasculaire (monocytes/macrophages résidents, cellules endothéliales, cellules musculaires lisses,...). Il en résulte notamment la synthèse et la sécrétion accrue de molécules contribuant au remaniement matriciel pariétal. Parmi les processus qui contribuent à la modification de la MEC, la glycation des protéines constitutives, et en particulier celle du collagène de la paroi artérielle, conduit à la formation et à l'accumulation de produits avancés de la glycation (AGEs) qui rigidifient le réseau fibrillaire. Les protéines de la MEC sont particulièrement visées par la glycation, en raison de leur très longue durée de vie. Les AGEs s'accumulent au niveau des lames basales vasculaires modifiant leur élasticité et leurs propriétés de filtration. En particulier, la glycation du collagène IV et de la laminine, qui sont des composants majeurs des lames basales, désorganise la structure matricielle, augmente leur durée de vie et favorise leur accumulation en rendant ces protéines modifiées résistantes à la protéolyse. Les AGEs exercent également leurs effets délétères par l'intermédiaire de leur interaction avec des récepteurs membranaires (RAGE). Cette interaction est responsable d'une cascade d'évènements biologiques qui, selon le type cellulaire, impliquent par exemple les voies de la NADPHoxydase, la p21Ras, les MAPkinases et les voies Jak/STAT et conduisent donc à la libération de cytokines pro-inflammatoires, de facteurs de croissance et de molécules d'adhésion [41]. Les AGEs formés dans la MEC pourraient également être à l'origine d'une inhibition de la synthèse de NO par les cellules endothéliales [42]. Une forme soluble de RAGE, agissant comme un leurre pour les AGEs en les inactivant en phase soluble, aurait un effet cytoprotecteur en assurant la neutralisation des AGEs sans conséquences cellulaires. Une concentration basse en RAGE soluble a été retrouvée chez des patients présentant des troubles cardiovasculaires et la gravité de l'atteinte semble inversement proportionnelle à cette concentration [42]. De telles modifications moléculaires de la MEC peuvent expliquer les anomalies du renouvellement de la matrice qui est physiologiquement assuré par le système MMP/TIMP [43,44].
Les MMPs (matrix metalloproteinases) sont des enzymes protéolytiques responsables du renouvellement permanent de la MEC de la paroi des artères saines. Plus de 25 MMPs ont été décrites chez l'homme. Les spectres protéolytiques de ces endopeptidases, initialement synthétisées sous forme de zymogènes, sont assez larges et se recoupent de manière importante. Ces enzymes peuvent dégrader la quasi-totalité des composants de la MEC. Néanmoins, elles peuvent être classées en six groupes selon les affinités pour les substrats, les analogies de séquence et l'organisation des domaines : matrilysines, collagénases, stromélysines, gélatinases, MMPs membranaires et MMPs atypiques. Pour permettre une adaptation fine de la protéolyse, l'activité des MMPs est régulée au niveau de leur expression et de leur activation extracellulaire. En ce qui concerne leur activité protéolytique, il existe des inhibiteurs physiologiques des MMPs : les TIMPs (tissue inhibitor of metalloproteinases). A la différence des MMPs, la famille des TIMPs, est plus restreinte : quatre principaux TIMPs identifiés (numérotés de 1 à 4) sont capables d'inhiber toutes les MMPs se liant avec le domaine catalytique des MMPs ou avec leur zymogène. Dans les conditions physiologiques, il existe un équilibre entre l'activité des MMPs et celle de leurs inhibiteurs. Ce réseau MMP/TIMP est fortement impliqué dans le remodelage du système cardiovasculaire et l'expression des MMPs et des TIMPs est modifiée avec l'âge : ainsi, MMP-2 et TIMP-1 ont une expression augmentée au cours du vieillissement, et l'augmentation de l'expression de MMP-9 est corrélée à une augmentation de la rigidité artérielle [45,46]. D'autre part, MMP-2 et MMP-9 qui ont toutes deux pour substrats l'élastine, certaines isoformes du collagène (IV pour MMP-2 et MMP-9, I, II, III pour MMP-2) et d'autres macromolécules de la MEC, sont impliquées dans la défaillance cardiaque post-infarctus du myocarde et l'apparition d'un anévrysme aortique. De plus, certaines MMPs pourraient intervenir dans le développement de la plaque athéromateuse [43,47]. TIMP-3 serait, quant à lui, impliqué dans le développement des cardiopathies avec dilatation [48]. Récemment, Bonnema et al. ont suggéré l'existence d'un lien entre le profil d'expression des MMPs et TIMPs, l'âge et la fonction ventriculaire gauche cardiaque, mais le mécanisme reste imprécis [49]. Enfin, un polymorphisme dans l'expression des MMPs, comme MMP-3 et MMP-9, pourrait expliquer, au moins partiellement la susceptibilité interindividuelle variable aux pathologies cardiovasculaires.
Les conséquences fonctionnelles artérielles des altérations de la MEC au cours du vieillissement et de l'HTA sont doubles : potentialisation du processus d'athérosclérose (le remodelage matriciel intimal étant un élément important de l'évolution des plaques athéroscléreuses [43]), et dégradation des propriétés élastiques de la paroi artérielle. A ce titre, la perte progressive de l'élastine au cours du vieillissement, par augmentation de l'activité élastolytique, a également été invoquée pour expliquer la perte de fonctionnalité de la MEC. La caractéristique métabolique de l'élastine est sa demi-vie très longue (supérieure à 40 ans) et l'absence de sa régénération lorsqu'elle est dégradée après les premiers mois de la vie. Une augmentation de l'activité élastolytique, par l'élastase notamment, est observée au cours du vieillissement et contribue à la dégradation de l'élastine qui n'est pas remplacée : les fonctions élastiques semblent alors être prises en charge de façon partielle par le collagène matriciel moins efficace, ce qui expliquerait en partie la contribution de l'élastolyse à la rigidification artérielle [50].
6.3 La calcification de la paroi artérielle
La calcification cardiovasculaire représente une marque de vieillissement, partiellement lié à l'exposition de sites de liaison du calcium sur l'élastine dégradée. Elle est également un stade évolué des plaques athéroscléreuses, observé fréquemment à un âge avancé de la vie (et parfois dès 60 ans) [50].
6.3.1 Les différents types de calcification artérielle
Classiquement, quatre types de calcification cardiovasculaires peuvent être identifiés :
- – la calcification athéroscléreuse : elle se produit au niveau des lésions athéromateuses/athéroscléreuses, donc au niveau intimal essentiellement. La calcification dans la lésion athéroscléreuse a comme point de départ la base des plaques de nécroses fibro-lipidiques via les vésicules apoptotiques provenant des CMLV mortes qui vont servir de support à des dépôts de phosphate de calcium. La calcification se développe de manière orientée et excentrique [50]. Elle débute dès le stade du noyau lipidique et prend la forme de lames osseuses histologiquement visibles. Il s'agit d'une calcification de type endochondral : son mécanisme semble similaire à celui de la calcification osseuse, associant une chondrogenèse préalable à une induction ostéoblastique. Cette calcification endochondrale est intimement liée à la néoangiogenèse du tissu athéromateux [51]. Dans l'athérosclérose, les structures ossifiées expriment des caractéristiques architecturales semblables à celles observées dans les os trabéculaires au niveau des zones de résorption ou de remodelage. Les lésions athéroscléreuses sont riches en monocytes-macrophages qui pourraient se transformer potentiellement en préostéoclastes dont la maturation nécessite la présence de RANKL et du facteur de différenciation des ostéoclastes (ODF). Les zones calcifiées seraient donc soumises à un processus analogue au remodelage osseux. Toutefois, la présence d'ostéoclastes proprement dits dans la paroi artérielle reste controversée même s'il existe des cellules qui en ont les caractéristiques [52].
- – la calcification média artérielle : caractéristique de l'artériosclérose et de la sclérose de Mönckeberg : il s'agit d'une calcification intramembranaire, l'étape de chondrogénèse n'étant pas nécessaire. La calcification de la MEC serait une conséquence, entre autre, de la migration à partir de l'adventice de myofibroblastes qui acquièrent un phénotype ostéoblastique sous l'action de facteurs tels que l'ostéoprotégérine (cf. infra) synthétisée par les CMLV [50]. Mais d'autre types cellulaires semblent impliqués. En culture, certaines populations de cellules musculaires de la média sont capables de produire une calcification. Ces cellules sont appelées cellules vasculaires calcifiantes. Elles peuvent se transformer en cellules proches des ostéoblastes et perdent alors leur capacité à exprimer les marqueurs spécifiques des CMLV. En transposant ce qui a été observé chez l'animal soumis à une hypervitaminose D3, la calcification pourrait être initiée par l'apparition de vésicules matricielles qui se développeraient autour des fibres élastiques, suivies par le dépôt de cristaux d'hydroxyapatite le long des lames élastiques et la dégradation de l'élastine. Une fois initié, le processus de calcification s'étendrait le long des fibres élastiques et aux CMLV environnantes. La calcification de la média artérielle peut avoir lieu en l'absence de macrophages ou de lipides [53].
- – la calcification valvulaire cardiaque : induite par le stress mécanique et l'inflammation qui se développe sur ce tissu, la calcification valvulaire apparaît moins structurée que les deux précédentes, dont elle combine les mécanismes [54].
- – la calciphylaxie vasculaire : il s'agit d'une artériolopathie de calcification de la media des artères de moyen calibre et des artérioles, associée à une prolifération intimale consécutive à une insolubilisation du phosphate de calcium en excès, le plus souvent au décours d'une insuffisance rénale. Cette calcification vasculaire particulière n'apparaît pas gouvernée par un processus ostéogénique [55].
Bien que différents, les mécanismes de la calcification de l'athérosclérose (endochondrale) et de la calcification médiale artériosclérotique (intramembranaire) font intervenir des acteurs cellulaires et moléculaires communs et possèdent des similitudes avec le processus d'accrétion osseuse [53]. Dans les deux cas, la calcification artérielle résulte à la fois d'une induction locale de l'ostéogenèse et d'un déficit d'inhibiteurs de la minéralisation. La principale hypothèse expliquant la calcification artérielle est une synthèse osseuse in situ par des cellules de phénotype ostéoblastique, issues de la transformation phénotypique des CMLV. Cependant, d'autres types cellulaires pourraient également subir une transformation ostéoblastique : les péricytes associés à l'endothélium et des cellules souches circulantes devenant résidentes au sein du tissu artériel.
6.3.2 Facteurs de l'induction de l'ostéogenèse
La transformation ostéoblastique des CMLV (et éventuellement des autres types cellulaires cités ci-dessus) s'opère sous l'influence de différents stimuli inducteurs de l'ostéogenèse, parmi lesquels les plus importants sont :
- – les acteurs du métabolisme phosphocalcique : (phosphates, parathormone, vitamine D). L'implication de ces éléments du métabolisme phosphocalcique et du remodelage osseux a été révélée par l'observation de calcifications artérielles chez des sujets atteints d'insuffisance rénale chronique. L'hyperphosphatémie engendrée chez ces sujets par une réduction de la filtration glomérulaire et un hyperparathyroïdisme secondaire, induit une diminution de la solubilité du phosphate de calcium, alors plus enclin à se déposer au niveau vasculaire et capable de favoriser la différenciation ostéoblastique des CMLV [56]. Le rôle de la parathormone et des peptides dérivés semble plus complexe et moins élucidé : ces molécules sont pro-ostéoblastiques au niveau osseux, en activant Cbfa1, mais inhibent l'évolution phénotypique pro-ostéoblastique des CMLV [57,58]. Par contre, l'action de la vitamine D et de ses dérivés (calcitriol) est clairement en faveur de la calcification artérielle, en induisant la différenciation phénotypique des CMLV et en augmentant leur activité phosphatasique alcaline, ainsi que l'expression du VEGF, de MMP-9, du collagène de type 1 et de l'élastine.
- – les bone morphogenetic proteins (BMP) : Ces molécules, appartenant à la superfamille des TGF-β, ont une activité ostéoblastique (BMP2, BMP4...) ou anti-ostéoblastique (BMP7) en interagissant, après liaison à leurs récepteurs (BMPR), avec les protéines transcriptionnelles Smads [59]. Les BMP ostéoblastiques, parmi lesquels BMP2 apparaît être le plus important, assurent la différenciation ostéogénique ou chondrogénique des différents types cellulaires. BMP2 est exprimé par les différents types cellulaires de la paroi artérielle, et son expression est favorisée par les mêmes stimuli que ceux favorisant l'activation des cellules endothéliales pariétales ou des CMLV (stress oxydant, turbulences hémodynamiques, hypoxie, lipides oxydés) [60].
- – système ostéoprotégérine/RANKL/RANK : Le récepteur RANK (receptor activator NF-kB) joue un rôle dans l'homéostasie osseuse en régulant l'ostéoclastogenèse après liaison à son ligand RANKL [61]. L'ostéoprotégerine (OPG) est un membre de la superfamille des récepteurs du TNFα. Sécrétée, elle sert de leurre pour RANKL et inhibe à la fois la différenciation cellulaire et le fonctionnement des ostéoclastes. Récemment, un lien entre la concentration plasmatique d'OPG et la sévérité des atteintes coronaires a pu être établi [62] : une élévation des concentrations plasmatiques d'OPG est associée à l'insuffisance coronarienne et à la mortalité cardiovasculaire et peut être considérée, au décours d'un infarctus du myocarde, comme un facteur de mauvais pronostic [63].
- – l'ostéopontine (OPN) : L'OPN est une phosphoprotéine retrouvée dans les tissus minéralisés. Elle semble jouer un rôle dans la calcification de la paroi des artères mais son rôle reste imprécis. Elle est absente dans les artères normales mais retrouvée en abondance dans les sites de calcification au niveau des plaques athéroscléreuses chez l'homme [64]. Certains travaux expérimentaux mettent en évidence plutôt un rôle délétère de l'OPN en particulier dans l'apparition de lésions athéromateuses majeures [65], alors que d'autre études plaident plutôt en faveur d'un rôle protecteur de l'OPN sur la paroi vasculaire. En effet, des souris n'exprimant pas l'ostéocalcine (MGP-/-) et dont le gène de l'OPN a été éteint (OPN-/-) présentent une calcification artérielle significativement accrue en comparaison de souris MGP-/- OPN+/+ [66].
- – les facteurs de transcription Cbfa1 et Ostérix : Comme dans la cas de l'accrétion du tissu osseux, l'activation de ces facteurs de transcription est responsable de l'expression de différentes protéines de la différenciation ostéoblastique : ostéocalcine, OPN, collagène de type I [67]. Cbfa 1 est également impliqué dans les interactions cellules-MEC médiées par les intégrines et, à ce titre, favorise l'action de BMP2 qui requiert de telles interactions pour exercer son action ostéoblastique. L'action cellulaire de Cbfa1 ne peut pas s'exercer sans un autre facteur de transcription, Ostérix, qu'il active : en retour, ce dernier semble alors agir de concert avec Cbfa1 pour exercer l'effet ostéogénique [68].
- – le stress oxydant : Un déséquilibre entre les agents prooxydants et antioxydants au profit des premiers est de nature à stimuler l'expression de Cbfa1 et BMP2 au niveau artériel. En effet, l'action de BMP2 semble passer par une voie redox-sensible, puisque cet agent induit l'expression de la sous-unité Nox1 de la NAD(P)H oxydase, et ainsi son activité. De façon plus générale, le stress oxydant peut être considéré comme un lien entre l'état inflammatoire et la calcification artérielle.
6.3.3 Facteurs de l'inhibition de la calcification
La seconde grande cause de calcification est la diminution des inhibiteurs endogènes de la calcification artérielle dont l'activité physiologique est de lutter contre la précipitation de complexes de calcium et de phosphate qui peut apparaître à des concentrations de calcium et de phosphate proches des valeurs physiologiques. Plusieurs types de molécules ont été décrits comme pouvant exercer cette action inhibitrice de la calcification, parmi lesquelles on peut retenir :
- – le pyrophosphate inorganique : Il est de plus en plus considéré comme un facteur paracrine d'inhibition de la calcification vasculaire. Les taux de pyrophosphate sont maintenus grâce à l'activité de la pyrophosphatase/phosphodiestérase 1 (NPP1) qui permet leur formation, et de l'ankyrine, qui assure leur externalisation. Un déficit de NPP1 ou de l'ankyrine est responsable d'une calcification artérielle accrue [69,70]. L'action du pyrophosphate pourrait être purement chimique, par inhibition de la formation de molécules d'hydroxyapatite très peu soluble, mais le pyrophosphate pourrait également inhiber la transformation ostéoblastique des CMLV. En tout état de cause, les études cliniques ont confirmé que l'étidronate, un analogue pharmacologique du pyrophosphate, assure une prévention de la calcification artérielle chez les sujets hémodialysés [71].
- – La gla protéine matricielle (MGP) ou ostéocalcine : Son action est complexe et éminemment liée à celle de BMP2. En fait, un ratio MGP/BMP2 inférieur à 1 ou supérieur à 15 semble favoriser la calcification, alors que des ratios intermédiaires l'inhibent [72]. La MGP peut être γ-carboxylée sous l'action de la vitamine K et former un complexe avec BMP2, ce qui inactive cette dernière et donc inhibe la calcification. Cette forme γ-carboxylée de la MGP est transportée dans le sang par la fétuine qui est une autre molécule inhibitrice de la calcification. La forme non γ-carboxylée est associée quant à elle à une calcification artérielle accrue : elle est retrouvée au niveau des zones lésées et calcifiées de l'arbre artériel [73].
7 Conclusion
Longtemps, l'artériosclérose a été considérée comme un vieillissement naturel des grosses et moyennes artères. On sait maintenant que c'est un phénomène pathologique qui n'est ni inéluctable, ni irréversible. Actuellement, les seules méthodes permettant d'évaluer l'état de l'arbre vasculaire s'appuient sur des examens cliniques ou des techniques d'imagerie. Or, l'artériosclérose est un processus éminemment biochimique dont chacun des acteurs peut être considéré comme un biomarqueur potentiel de l'état de l'arbre vasculaire. Il apparaît aujourd'hui important d'évaluer l'apport du dosage de molécules témoins ou actrices du dysfonctionnement endothéliale et/ou de la calcification vasculaire dans le contexte du vieillissement de l'arbre artériel. L'identification de biomarqueurs du processus artérioscléreux pourra alors compléter ceux utilisés actuellement pour le processus athéroscléreux (marqueurs lipidiques notamment) en vu d'une évaluation biologique précise et utile du risque vasculaire au cours du vieillissement.