

1 Introduction
Conservation biology is a relatively new field of research that emerged in the seventies with the goal of preserving ecosystems, species and genes [1]. Within this field, conservation genetics deals with the application of genetic concepts and tools to conservation problems. At first glance, highlighting conservation problems in domestic ungulates may appear as a paradox [2], as the census population sizes of farm animals are extremely high (Table 1). Potential conservation issues in domestic animals were already spotted by the Food and Agriculture Organization of the United Nations (FAO) in the seventies [3] with the objective of long-term conservation of genetic resources for future use. Such a goal can be achieved by preserving the widest possible spectrum of genetic diversity because future needs are unpredictable. This FAO document [3] also emphasized the conflict between immediate improvement and conservation, indicating that “the best way of conservation would be the development of a management system which would both maintain genetic variability of existing livestock resources and at the same time permit continuous improvement in productivity and adaptability of that resource”. Unfortunately, despite this very early warning, the investments toward conservation aspects were far behind those for improving the productivity of a few industrial breeds. As a consequence, traditional indigenous breeds are still disappearing (Table 1) and the effective population size of many cattle breeds is far below a threshold that would ensure long-term sustainability. The last FAO survey [4] considered that 16, 13 and 3% of the cattle, sheep, and goat breeds already disappeared, respectively. In such a context, it is becoming more and more urgent to implement sound conservation strategies for farm animals [5].
Population sizes, current number of breeds, and number of extinct breeds for cattle, sheep, and goats at the worldwide level (statistics concerning 169 countries [4]).
| Cattle | Sheep | Goat | |
| Population size (’000) | 1,367,335 | 1,060,606 | 710,381 |
| Number of breeds | 1311 | 1409 | 618 |
| Number of extinct breeds | 209 | 180 | 19 |
In this paper, we first aim to review the current knowledge about the origin and the history of cattle, sheep, and goats. Such knowledge is of prime importance for properly assessing the risk concerning the loss of genetic diversity and for designing sound conservation guidelines. We also examine the current practices that lead to massive loss of genetic resources. Then, we highlight the potential genetic resources, obviously including domestic breeds but also including wild relatives. Finally, we provide conservation guidelines for long-term sustainability based on the current situation and on the availability of new genomic tools.
2 The origin of cattle, sheep and goats
Data about cattle, sheep, and goat domestication first came from osteometry and morphometry evidence collected in archaeological sites [6]. More recently, these data were completed by extensive genetic studies [7] using both modern and ancient samples, thus allowing producing more precise scenarios of the domestication processes. The first archaeological evidence of the domestication of these three species traces back to around 10,500 BP (calibrated) in the Fertile Crescent (Fig. 1). It seems that goat and sheep were domesticated first [8,9], immediately followed by cattle [10].
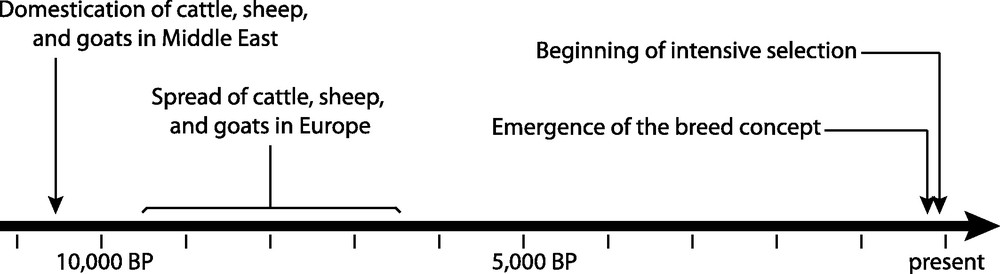
The main events in cattle, sheep, and goat domestication.
The wild ancestor of all domesticated cattle was the auroch (Bos primigenius) [11] that is now extinct. Aurochs had a very wide geographic distribution, from East Asia to Europe and North Africa. The common usage accepts two taxa for the domestic cattle, namely Bos taurus and B. indicus that fully interbreed. B. indicus differs from B. taurus by the presence of a prominent hump. The presence of two mtDNA haplogroups (Fig. 2) is interpreted as an indication of two main domestication events, the one in the Fertile Crescent leading to B. taurus and the other in the Indian subcontinent leading to B. indicus [12–14]. Extensive hybridization occurred in Africa, leading to a complex intermixing of these two mitochondrial haplogroups in the field [13].

Unrooted neighbor-joining trees illustrating the mtDNA polymorphism of cattle, sheep, and goats [2,51].
The systematics of the genus Ovis is controversial. The number of species that have been recognized within this genus varies from one [15] to seven [16]. Recently, an extensive mitochondrial and nuclear DNA survey including all taxa of the genus was carried out [17], confirming the presence of seven monophyletic clades corresponding to the seven species described by Nadler et al. [16]. Based on another extensive study of the mitochondrial DNA (mtDNA) of putative ancestors (O. orientalis, the Asiatic mouflon; O. vignei, the urial; O. ammon, the argali), it clearly appeared that the wild ancestor is O. orientalis [18]. Both archaeological [8] and genetic data spot the domestication center in eastern Anatolia and North-west Iran. To date, the number of mtDNA haplogroups described for the domestic sheep varies as no standard criteria are used for defining them [19,20]. However, authors agree on three main haplogroups (i.e. A, B and C in Fig. 1).
Goat domestication is well documented by archaeological evidence and also by several genetic studies. The goat wild ancestor is the bezoar, Capra aegagrus [21–23]. A large-scale analysis of current bezoar mtDNA polymorphism over its whole geographic distribution has recently been performed [23]. All the six current mitochondrial haplogroups found in the domestics (Fig. 2) were also found in its wild ancestor, suggesting that the domestication process occurred over a very large area encompassing eastern Anatolia and North-West Iran.
The cattle mtDNA polymorphism [12,13,24] seems compatible with the domestication of only two mtDNA haplotypes, leading to B. taurus and B. indicus. Such an observation is in favor of a population bottleneck at the domestication time. On the opposite, the goat mtDNA polymorphism of the A haplogroup (representing more than 90% of the haplotypes) is by far too high for originating from a single haplotype at the domestication time. A detailed analysis of this A haplogroup suggested an initial number of haplotypes higher than 1000 [23], strongly supporting the absence of bottleneck at the domestication time in goats. The sheep mtDNA polymorphism also seems to support an absence of bottleneck. How can the differences between cattle on one hand, and goat and sheep on the other hand, be explained? If we assume that a sustainable domestication must be based on a very high gene pool, we may elaborate the following scenario. As the geographic distribution of sheep and goat wild ancestors was quite restricted around the domestication centres, only a large-scale domestication event involving thousands of individuals can produce a population that will not suffer from inbreeding depression later on, during the dispersal out of the domestication centres. The cattle domestication might have followed a different trajectory, with first a bottleneck at the domestication time, followed by extensive introgressions from the aurochs later on, over large geographic areas as the wild ancestor was very widely distributed [25]. Such introgressions were able to enlarge the nuclear DNA gene pool of cattle until the extinction of the aurochs in the sixteen century. We can also imagine that these introgressions were strongly biased toward male aurochs mating with domestic females, explaining why cattle mtDNA only transmitted by female does not show significant auroch contribution. However, aurochs’ mtDNA have been found at a very low percentage in Italian cattle breeds [26,27]. Recent studies [28] suggest that introgressions from aurochs into domestics may have been even more widespread than expected. This might partly explain the relatively large cattle gene pool despite a likely bottleneck at the time of domestication.
3 Dispersal from the domestication centres
During the 3000–4000 years following the initial domestication events in the Fertile Crescent, the Neolithic culture diffused over Europe, Africa and Asia. Archaeological evidence showed that agriculture colonized Europe by two main routes, the Mediterranean route and the Danubian route. Due to successive founder effects during the spread of agriculture, we can hypothesize a clinal decrease in genetic diversity with increasing distance to the domestication centre. Such a decrease has been demonstrated for cattle mtDNA, for which populations in Western Europe exhibit lower polymorphism than those in the Near East [14,24]. This might not be initially the case for cattle nuclear DNA if introgressions from aurochs were common. However, the pattern of variation observed may also be the result of recent selective processes; it has been recently shown that traditional cattle breeds closer to the domestication centre in Middle East show a higher nuclear polymorphism than more heavily selected breeds in Western Europe [29]. Several secondary migrations accompanied human migrations in more recent historical times and contributed to a more complex shaping of local gene pools. For example, it has been shown that Iberian cattle breeds have been introgressed by African breeds [30–33]. Also, a close genetic relationship was discovered between Tuscan cattle breeds and Near Eastern breeds, possibly linked to the sailing and docking in Tuscany of Middle Eastern people in the late Bronze Age and to the onset of the Etruscan civilization in Central Italy [34]. Surprisingly, despite potential serial founder effect during the agriculture spread, the nuclear DNA polymorphism based on microsatellites is still relatively high in the three species [35–38], comparable to what is found in wild species. Such a result suggests a large effective population size during most of the period since the colonization.
4 Influence of the breed concept on genetic diversity
Fig. 1 summarizes the history of cattle, sheep, and goats. During about 10,000 years, farmers controlled the reproduction of their farm animals by favoring the reproduction of individuals with better phenotypes. As a result, farm animals slowly became adapted to local environments and fulfilled the needs of farmers in a sustainable way. At that time, gene flow among different phenotypes was possible, resulting in relatively high effective population sizes, preventing genetic drift at the regional scale. The situation dramatically changed about two hundred years ago with the emergence of the breed concept. Since that time, much stronger selection pressures have been applied to local populations followed by standardization of the morphology and the performance. All animals from the same breed began to exhibit the same phenotypic characteristics, including the same coat color. Most importantly, the gene flow among different phenotypes (i.e. among different breeds) was seriously reduced. To summarize, as illustrated on Fig. 3, the populations of farm animals that underwent relatively soft selection pressures during about 98% of their common history with humans were suddenly fragmented into many well-defined breeds, with much higher selection constraints. Population fragmentation is known to have deleterious consequences in the long term by increasing genetic drift and inbreeding, and by reducing the fitness [39]. Fragmentation is also one of the most important factors leading to extinction in wild species [39].

Different phenotypes in the past now replaced by a homogenous breed.
5 The current loss of genetic resources
The current loss of genetic resources concerns not only the extinction of traditional breeds, but also the loss of genetic diversity within breeds. With the development of artificial insemination during the last 50 years, only a few males were involved in reproduction schemes and consequently industrial breeds underwent another important step toward the reduction of their effective population size. For example, at the worldwide level, the Holstein cattle has an effective population size of about 50 (Table 2), leading inexorably to genetic drift and loss of alleles. All the genetic diseases observed in this breed [40], as well as the strong reduction in fertility [41] may be linked to such a low effective population size. Based initially on data from livestock, conservation biologists proposed the 50/500 rule of thumb for managing wild species [42]: in the short term, the effective population size should not be less than 50 to avoid extinction risk due to genetic effects; in the long term, the effective population should not be less than 500. Surprisingly, such low effective population sizes in many cattle industrial breeds did not warn scientists in charge of the management of these breeds, maybe because a low effective population size is an advantage for short-term genetic selection and performance improvements.
Traditional breeds are threatened by the success of industrial breeds via two processes. First, the high performance of industrial breeds tends to impose the replacement of traditional breeds by more productive ones. In many areas, farmers have a strong economic pressure to switch to industrial breeds. Such a phenomenon can be very fast, and a valuable traditional breed can be lost within a decade. Second, autochthonous breeds are often crossbred to a more productive breed from elsewhere, most often a high production breed. Adaptive traits may be rapidly lost by poorly designed crossbreeding leading to dilution of important adaptive loci of traditional breeds. Traits such as resistance to local infectious and parasitic diseases, adaptation to poor forage, homing and gregarious behavior can be rapidly lost and difficult to rescue. They represent key traits for the survival and the management of herds in extensive farming. In developing countries, many examples illustrate this introgression threat, where indiscriminate repeated crossbreeding quickly disrupted generations of selection for adaptation to harsh environments (see examples in [2]).
6 The potential genetic resources
A basic approach for characterizing these resources can simply be to record phenotypic characters in all the very diverse breeds adapted to different environments and management systems. Then the genetic resources can be identified based on these phenotypic characters, assuming that phenotypes are strongly linked to genetics. An alternative approach consists to employ genetic markers. However, the characterization of genetic resources with molecular markers suffers two difficulties: the ascertainment bias of the markers used, and the problem of neutral versus adaptive markers.
The ascertainment biases are due to the adjustment of genetic markers (microsatellites or single nucleotide polymorphisms) with the constraint of being as polymorphic as possible in the breeds under study, i.e. industrial breeds in most cases. As a consequence, the genetic diversity estimates for traditional breeds when using such markers will be biased toward low values and the estimation of heterozygosity is particularly affected [43]. Such an ascertainment bias has also been clearly shown in wild species where only the most polymorphic microsatellites are selected for further investigations [44]. Genome-wide analyses based on single nucleotide polymorphisms (SNP) are now widely used in farm animals [45], but also suffer from the same ascertainment bias [46,47]. In such a situation, only sequencing many regions of the genome would give a reliable estimate of the genetic diversity [44] and is appropriate for properly characterizing genetic resources.
The second difficulty for characterizing genetic resources is linked to an ongoing debate about the relative importance of neutral versus adaptive variations for identifying the populations to prioritize for protection purposes. The neutral variation, an indicator of global genetic diversity, can be used for assessing conservation values, based on the idea that we do not know future selection pressures and that more diverse populations at the genome-wide level will better adapt [48]. The opposite point of view claims that only adaptive variation is relevant for conservation purpose [49].
Should we include wild ancestors as potential genetic resources when they still exist? The answer to this question is not straightforward, as no extensive study has been carried out so far to properly assess the potential of wild populations as genetic resources. The value of wild ancestors will be inversely linked to the proportion of genetic diversity that has been captured during the domestication process. Within each of the Capra and Ovis genera, many species can hybridize and produce fertile offspring. As a consequence, beside the wild ancestor, several other species within these two genera can also be considered as genetic resources (e.g. Capra falconeri for goats, Ovis vignei and O. ammon for sheep; Table 3). For cattle the situation is different, as the wild ancestor is extinct. Nevertheless, four or five wild species of the genus Bos are still alive and can produce fertile hybrids with cattle. Thus, they might also be considered as genetic resources (Table 3).
Wild species representing potential genetic resources for cattle, sheep, and goats (only the most closely related species are presented for sheep and goats).
| Common name | Scientific name | Geographic distribution | Domestic form | Conservation status [56] |
| Gaur | Bos gaurus | South and southeast Asia (largest populations in India) | Bos frontalis | Vulnerable |
| Banteng | Bos javanicus | South Asia (Java, Borneo, Myanmar, Thailand, Cambodia, Laos, Vietnam) | Bali cattle | Endangered |
| Kouprey | Bos sauveli | Northern Cambodia, southern Laos, Western Vietnam, eastern Thailand | – | Critically endangered (possibly extinct) |
| Yak | Bos mutus | Himalayan region of south Central Asia (Tibetan Plateau, Mongolia, Russia) | Bos grunniens | Vulnerable |
| Asiatic Mouflon | Ovis orientalis | Caucasus, northern Iraq, northwestern Iran, Anatolia | Ovis aries (sheep) | Least concern |
| Urial | Ovis vignei | Asia minor | – | Vulnerable |
| Argali | Ovis ammon | Central Asia | – | Near threatened |
| Bezoar | Capra aegagrus | Central Afghanistan, southern Pakistan, Iran, western Turkmenistan, northern Iraq, Caucasus region, Turkey | Capra hircus (goat) | Vulnerable |
| Markhor | Capra falconeri | Northeastern Afghanistan, Gilgit-Balistan, Hunza-Nagar Valley, northern and central Pakistan, Kashmir, southern Tajikistan, southern Uzbekistan | – | Endangered |
7 Conclusion
The effective management of farm animal genetic resources is primordial to ensure global and sustainable food security. The erosion of genetic resources has been clearly documented for farm animals [4]. Within a few decades, we might lose most of the highly valuable farm animal genetic resources that humanity has gradually selected over the past 10,500 years [2]. Urgent conservation measures must be taken to avoid such an irremediable loss [5]. These genetic resources are even not properly characterized due to the problem of ascertainment bias of the molecular markers used up to now. Fortunately, with the development of the next generation DNA sequencing technology [50], it will be possible to resequence whole genomes and to properly assess the genetic diversity and the conservation value of the different breeds, avoiding the ascertainment bias due to the use of microsatellites or single nucleotide polymorphisms. There is clearly a race between the characterization of genetic resources and their loss. In the same way, the development of genomic tools will allow to optimize the breeding strategies for ensuring the improvement of performance together with the preservation of genetic diversity.
If the integration of wild relatives in conservation planning is common in plants, it is not the case for domestic animals. Most of the wild relatives of cattle, sheep, and goats are endangered (Table 3), and no ongoing actions were implemented to preserve them based on the fact that they have the potential of representing valuable genetic resources for agriculture. Thus, it is now urgent to properly assess their potential as genetic resources.
The first step toward an efficient conservation strategy for cattle, sheep, and goat genetic resources is the proper characterization of the conservation value of the different breeds and of the wild relatives. This step relies on genetic technologies, and we can be optimistic at that level according to the current revolution in DNA sequencing. However, the implementation of the subsequent steps is more puzzling, as conservation strategies for farm animal genetic resources must integrate economical, sociological, and political parameters.
Conflict of interest statement
The authors have not declared any conflict of interest.
Acknowledgements
The European Commission provided funding via the FP7 NextGen project (“Next generation methods to preserve farm animal biodiversity by optimizing present and future breeding options”; Grant agreement no. 244356).