

1 Introduction
Noncoding RNAs (ncRNAs) form an important class of macromolecules participating in key cellular mechanisms such as protein synthesis, gene splicing, telomere elongation, regulation of gene expression (e.g., riboswitches, miRNAs, and other small regulatory RNAs), and gene silencing. In general, ncRNAs are specific transcripts that often participate in complex regulatory mechanisms [1–3].
Finding ncRNAs in genomes is far from trivial. First, the ncRNA genes lack the characteristic features of protein genes such as start and stop codons, splicing sites, codon frequency bias [4]. Second, structured ncRNAs are, in general, more conserved in structure than in sequence due to base covariations in helices and neutral substitutions in tertiary interactions [5,6]. Third, insertions of long sequences occur frequently [7–9]. These characteristics reduce the effectiveness of searches based on pure sequence comparison. Finally, the curation of ncRNA gene predictions is time consuming and demand expertise. In spite of these difficulties, the increasing recognition of the importance of ncRNAs in biological processes makes their annotation an essential component of any genome sequencing project.
To guarantee a timely and effective annotation of ncRNAs, multi-genome sequencing projects, such as the Génolevures project [10], require, as far as possible, automatic gene annotation and a set of simplifying procedures and tools for fast and accurate manual curation.
The problem of genomic ncRNA annotation has been tackled by several authors [11–21]. However, the specificities of each project, such as the differences in ncRNA biology between species, genome size, sequence and structure divergence, demand a careful analysis before applying any known method.
Here, we present our approach of ncRNA annotation in the context of the Génolevures consortium. We assembled a pipeline integrating publicly available tools for ncRNA search in sequences and applied it to the annotation of 9 budding yeast genomes from the Génolevures Database [22] and the Ashbya Genome Database [23]. We were able to annotate automatically, with a relatively small human validation effort, 693 ncRNAs that correspond to 81% of what we would expect taking the 86 annotated ncRNAs of the Saccharomyces cerevisiae genome as a reference.
2 Results
2.1 Homology pipeline
Homology search consists in searching for new members of known ncRNAs families. The important, and increasing, number of publicly available ncRNA annotations makes homology search the first step in any ncRNA annotation process.
Search tools based on sequence alignment, such as BLAST [24], allow for very fast searches of sequences (query sequences) in genomes (target sequences). The sequence similarity between query and target sequences plays a major role in the success rate of BLAST searches. Significant candidates (i.e. those with E-values < 0.1) will present sequence similarities above 84% with a minimum length of 16 nts. Thus, the use of BLAST for homology search becomes more effective for ncRNA families with large conserved sequences, such as rRNA or snRNAs, or when searching within closely related species. When the target and query sequences come from distant species, or present low sequence conservation, with potentially large insertions, pure sequence alignment methods loose efficiency. A way to get around this limitation is to explore the statistical signals imprinted on the sequence by the RNA structural constraints.
ncRNAs present complex three-dimensional structures of packed double-stranded helical regions connected by tertiary interactions that are generally mediated by single-stranded regions [25]. The set of double-stranded and single-stranded regions is called secondary structure. RNA helices consist of stacks of A–U; G–C and G–U base pairs. Those base pairs are structurally equivalent, and the substitution of one base pair by another one has, frequently, minimal or no impact on the molecular structure. The accumulation of base pair substitutions in a RNA sequence can render two homologous sequences very dissimilar. However, when observed from the point of view of a multiple sequence structural alignment, it generates a pattern of covariation between the base-paired positions that can be detected in the respective columns of the sequence alignment [26,27]. This dependency between paired positions in alignments is used to build covariance models, used by ncRNA search tools such as INFERNAL [28], to search for ncRNAs in large sequences. Additionally, if sufficiently diverse sequences are included in the alignments, the known positions of insertions can also be included into the models. Covariance models, for most of the known ncRNAs families, are curated and maintained in the RFam database [29] and could be readily applied in our search.
The results produced by any search tool must be automatically filtered in order to exclude candidates less likely to be real ncRNAs. Candidates conflicting with known annotations or with low score should be discarded from the candidate list. Additionally, all retained candidates should be structurally aligned with known homologues in order to facilitate human validation, required as the last step of the annotation process (Fig. 1A).
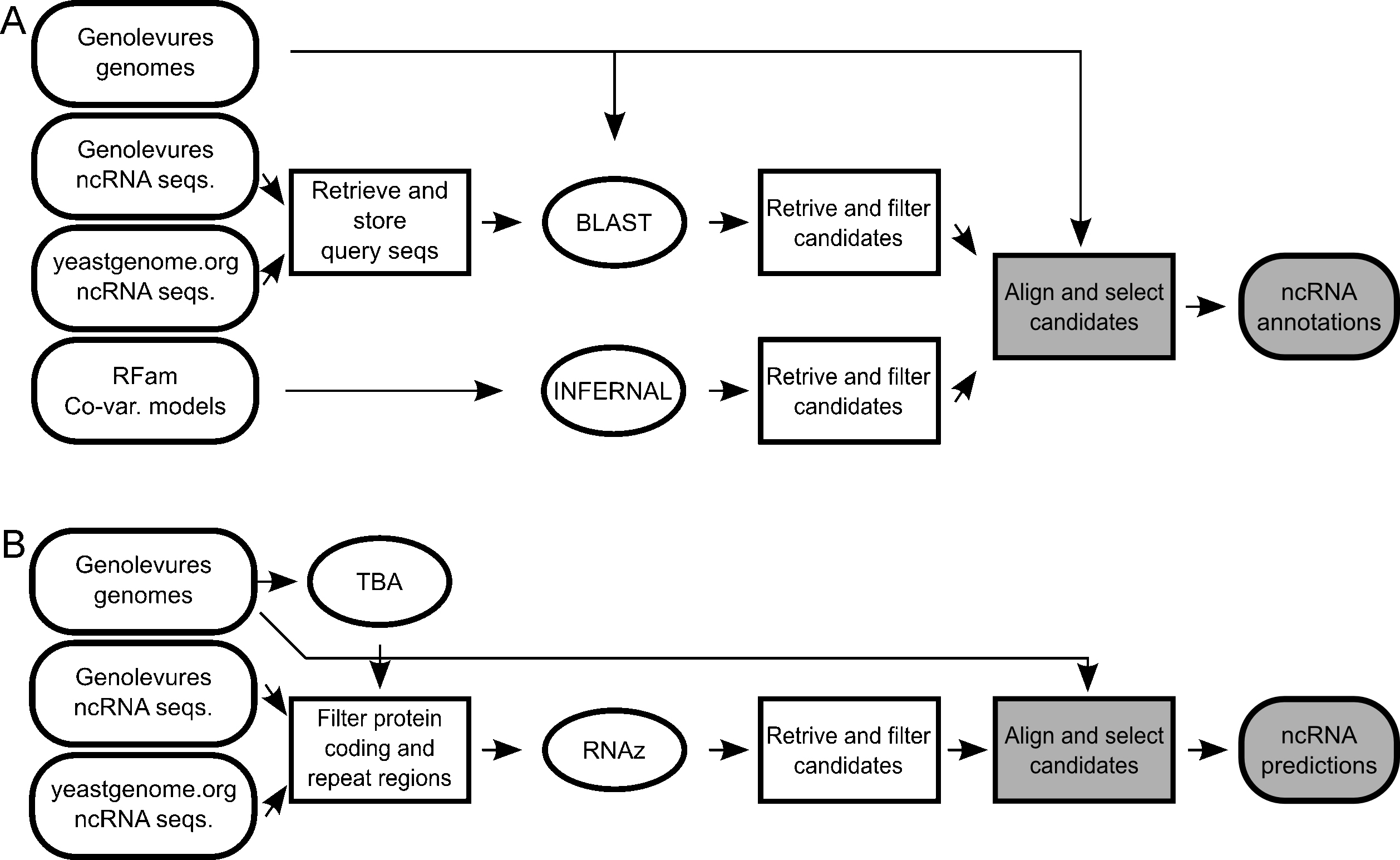
Workflow of the two pipelines for the ncRNA search and annotation. The pipelines integrate external sources of data (round white squares), external tools (ellipsis), automatic (white squares) and manual (shaded squares) processing steps to produce final ncRNA predictions and annotations (round shaded squares). A. Homology search pipeline. B. De novo search pipeline.
The Génolevures database [22] contains 9 budding yeast genomes in which only tRNAs and rRNAs were fully annotated and were not considered in this work. Some other ncRNA families were partially annotated (see Table 1, column “Original Annotation”). The Ashbya Genome Database [23] contains the fully annotated genome of the Ashbya Gossypii (also known as Eremothecium gossypii) yeast.
Number of ncRNAs found with the homology pipeline. Rows contain species numbers, columns contain numbers for each ncRNA family. First row (sace) displays the reference number of annotated ncRNA in the Saccharomyces cerevisiae genome. Original annotation column refers to ncRNAs annotated previously to the present work. “Sace specific” column refers to 4 ncRNA annotated in the S. cerevisiae genome.
| Species | Original Annotation | Rnase P | SRP | Rnase MRP | Telomerase | snRNA | snoRNA C/D | snoRNA H/ACA | Sace specific | TOTAL |
| sace | 86 | 1 | 1 | 1 | 1 | 5 | 44 | 29 | 4 | 86 |
| Number of found ncRNAs | ||||||||||
| cagl | 9 | 1 | 1 | 1 | 0 | 5 | 42 | 24 | 0 | 74 |
| zyro | 48 | 1 | 1 | 1 | 0 | 5 | 43 | 23 | 0 | 74 |
| sakl | 50 | 1 | 1 | 1 | 0 | 5 | 44 | 25 | 0 | 77 |
| klth | 49 | 1 | 1 | 1 | 0 | 5 | 44 | 24 | 0 | 76 |
| klla | 50 | 1 | 1 | 1 | 0 | 5 | 44 | 24 | 0 | 76 |
| ergo | 75 | 1 | 1 | 1 | 0 | 5 | 42 | 24 | 0 | 74 |
| deha | 8 | 1 | 0 | 0 | 0 | 5 | 39 | 17 | 0 | 62 |
| piso | 0 | 1 | 0 | 1 | 0 | 5 | 35 | 17 | 0 | 59 |
| yali | 8 | 1 | 0 | 1 | 0 | 5 | 32 | 13 | 1 | 53 |
| arad | 0 | 1 | 1 | 1 | 0 | 5 | 33 | 14 | 0 | 55 |
| Total | 297 | 10 | 7 | 9 | 0 | 50 | 398 | 205 | 1 | 680 |
We performed a BLAST search on those 10 genomes using as queries the S. cerevisiae ncRNAs and all the originally annotated ncRNAs from the original databases. This set of query sequences corresponds to the closest species for which we had reliable and ready to use ncRNA annotations. We believe that using a larger and more distant set of ncRNA sequences would increase the low score candidates with little improvement on the amount of genes found (e.g. of the 86 S. cerevisiae query sequences 37% produced true positive candidates when BLASTed against Candida glabrata, but only 6% when BLASTed against the more distant Debaryomyces hansenii and Yarrowia lypolitica–see Table SI 1). The BLAST search produced 1540 candidates with E-values < 0.1. A first INFERNAL search, using the 83 covariance models corresponding to the ncRNA families present in S. cerevisiae genome, produced 1250 candidates with E-values < 0.5. The candidates were included in the structural alignment of the corresponding family and manually validated according to the criteria described in the section “Candidate Validation”. After validation we retained 554 BLAST candidates and 602 INFERNAL candidates as ncRNAs.
We were interested on how E-values behave as classifiers in this specific data set. Fig. 2 shows the ROC curve [30], after the manual classification of True and False positives (TP and FP), for both tools. INFERNAL E-values were slightly better discriminators than the E-values of BLAST. While both tools will achieve 99% of True Positive Rates (TPR) at similar E-values (0.07 and 0.08 for INFERNAL and BLAST, respectively), the False Discovery Rates (i.e., the proportion of FPs in selected candidates) is significantly lower for INFERNAL (0.35) than for BLAST (0.61).
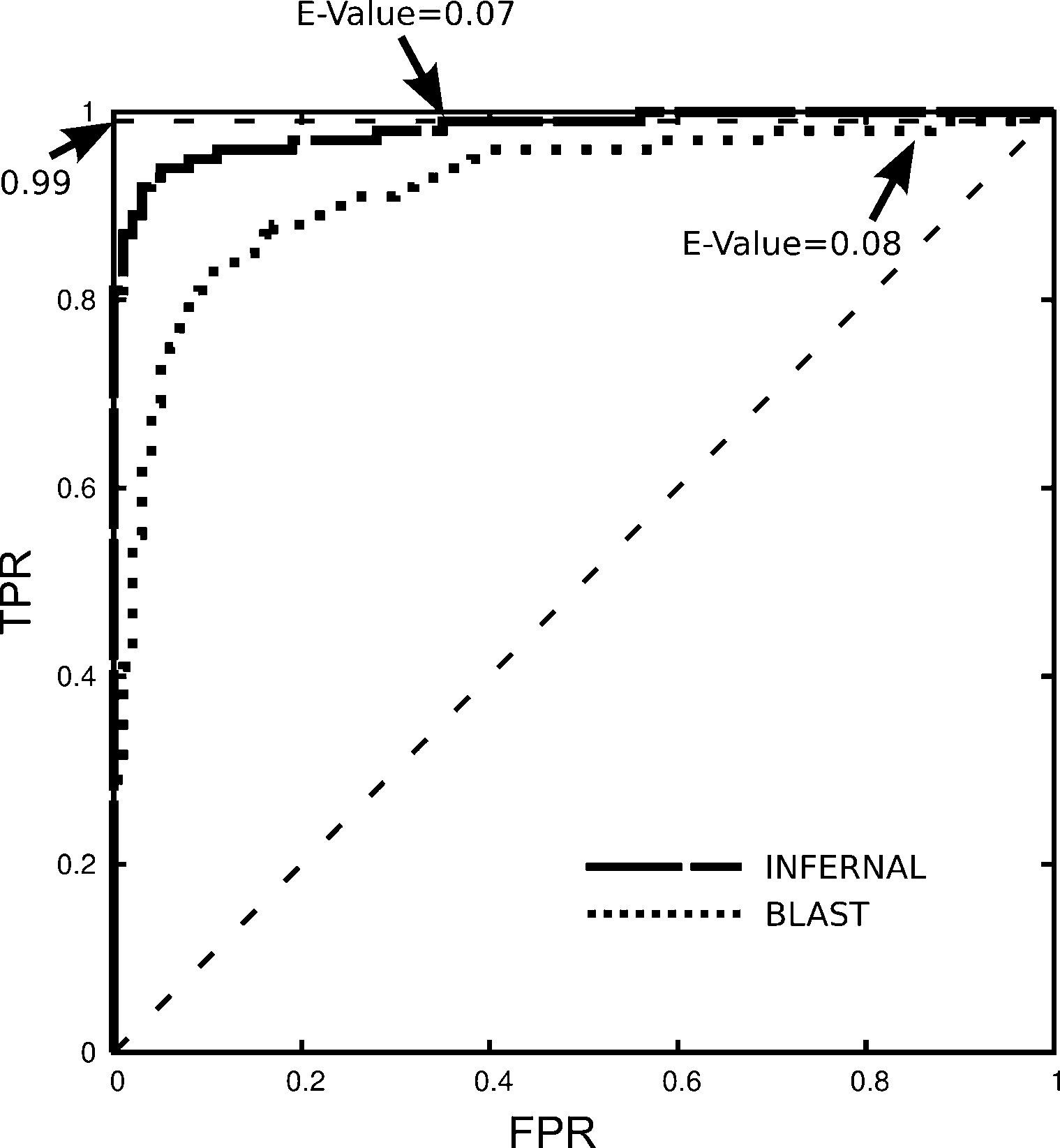
Receiver Operating Characteristic (ROC) curves for INFERNAL (dashed curve) and BLAST (dotted curve) E-values. INFERNAL E-values are slightly better discriminants (Area Under the Curve [AUC] = 0.98) than BLAST E-values (AUC = 0.92). A True Positive Rate (TPR) of 0.99 implies a False Positive Rate (FPR) of 0.34 for INFERNAL (E-Value = 0.07) and 0.87 for BLAST (E-Value = 0.08).
A second INFERNAL search was performed including all the remaining RFam families with exception of viral and miRNA families (719 Rfam families). According to the sensitivity analysis performed previously (Fig. 2), we reduced the expected value cutoff to < 0.1 obtaining 1360 candidates (applying the less restrictive cutoff, E-values < 0.5, we would have obtained 5578 candidates). From this search, only 41 candidates were selected. This surprisingly low number can be explained by the fact that most of the 719 Rfam families do not have homologues in yeasts, thus the resulting candidates are mostly FPs with high E-values (see Supplementary material SI 1). Curiously, all except two of the selected candidates correspond to homologous ncRNAs not found in the first search. A representative example is the box C/D snoRNA snR47 that was identified in Y. lypolitica, D. hansenii, K. thermotolerans and Z. rouxii with the covariance model of the snoRNA SNORD36 that is the mammalian ortholog of snR47. The remaining two candidates were, until now, unknown in the Saccharomycotina clade: a Box C/D snoRNA found in Y. lypolitica (Fig. 3) and a TPP riboswitch found in the 5’ UTR of protein coding genes in three different species (Fig. 4). The snoRNA candidate was found by INFERNAL using a covariance model of an archaeal snoRNA from the Pyroccocus family. The conservation of the characteristics Box C/D, and the complementarity of the putative guide sequences with the ribosome prevent a rapid exclusion of this candidate. On the other hand, the fact that the homologues of the putative ribosomal target sites are not modified in S. cerevisiae raises doubts about the nature of this candidate that only experimental validation could confirm. The TPP riboswitches are the only riboswitches found in eukaryotes [31,32] and the structural alignment with the crystal structures of the bacterial (Escherichia coli) [33] and plant (A. thaliana) [34] TPP riboswitches reveals a striking similarity of key structural nucleotides. The distribution of this regulatory domain across the Saccharomycotina phylogeny (Fig. 4B), totally absent in the S. cerevisiae branch, while present in D. hansenii, A. adeninovorans. and Y. lypolitica, raises interesting questions about the evolution of this type of mechanisms, and show the utility of extending homology search beyond the close group of species.
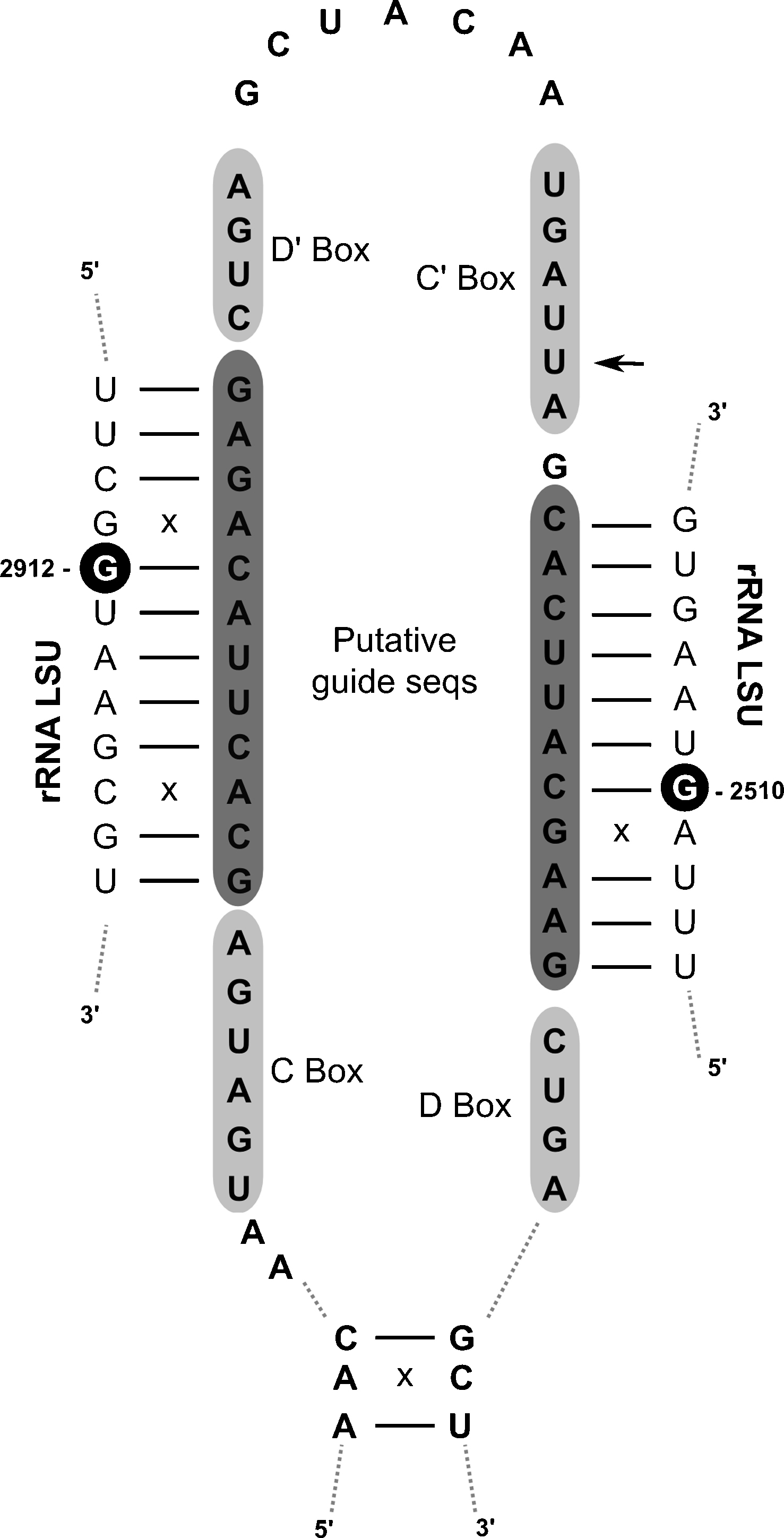
Secondary structure of the C/D box snoRNA candidate found in Y. lypolitica. The size, sequence and position of the putative C/D and C’/D’ boxes are compatible with a typical C/D snoRNA (black arrow points to a deviation from a canonical C’ Box). The putative guide sequences are complementary to two regions of the Ribosomal Large Subunit of Y. lypolitica (with pairs missing Watson-Crick complementarity indicated by small ‘x’). The predicted modified positions (white nucleotides in black circles) are not known to be modified in yeast.

TPP riboswitch candidates found by homology. A. Multiple structural sequence alignment of the seven predicted candidates. The ‘ + ’ and ‘*’ indicate columns conserved in at least 90 and 99% of known sequences respectively. In bold are the nucleotides deviating from the consensus. The last row represents the secondary structure in bracket notation. Candidate sequences are compatible with the observed sequence conservation and secondary structure. B. Phylogenetic tree of the searched species. Stars represent the number of TPP ncRNAs found in each organism.
Table 1 presents the results of the homology pipeline distributed by species and ncRNAs families. The total retained candidates correspond to 79% of all ncRNAs that were expected assuming the S. cerevisiae database as the reference for the ncRNA families present on yeast. At least 60% of the expected ncRNAs were found in all species. Unsurprisingly the homology search was much more effective in the species from the upper branch (from S. cerevisiae to A. gossypii), as 90% of the sequences used as queries came from that branch. Comparing the proportion of ncRNAs found by INFERNAL and BLAST for each species (Fig. 5B), we can observe that BLAST was as sensitive as INFERNAL as long as the searched genomes were close enough. In more distant species, while both tools loose performance, INFERNAL is much more sensitive.
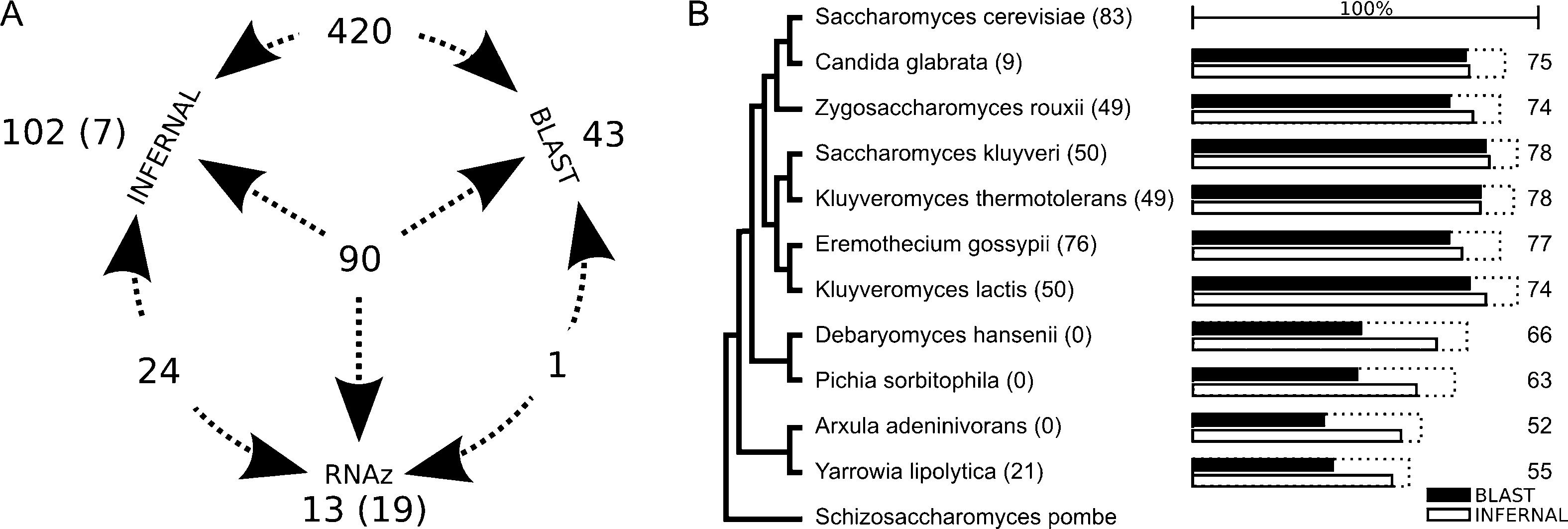
Number of ncRNAs found using each of the tools. A. The numbers beside each tool name correspond to ncRNAs found exclusively by that tool. Numbers between two arrows are ncRNAs found by both tools. The number of ncRNAs found by the three tools is at the center. In parenthesis are the found ncRNAs with no homologue in Saccharomyces cerevisiae. B. Proportion of ncRNA found in each genome by INFERNAL and BLAST. While both tools decrease performance with increasing evolutionary distance, INFERNAL is less affected than BLAST. Numbers in parenthesis indicate the number of ncRNAs originally annotated and used in the homology search. Dotted squares and numbers correspond to the total proportion and absolute number of found ncRNAs after running both pipelines.
Finally, it was not possible to find the RNA component of the Telomerase complex with any of the used tools. This ncRNA presents a challenge to automatic search programs due to minimal sequence and secondary structure conservation, extensive insertions/deletions and variable size between species [9,35,36]. The telomerase ncRNAs contains some structural features that are common to most known yeasts [36] such as the template region complementary to the Telomeric Repeat Sequence (TRS), a characteristic uridine-rich pseudoknot, two helical regions known to be the binding sites of the yKu and EST1 complexes, and a uridine-rich Sm binding domain. The template region can be detected with a simple BLAST using the TRS of the organism as a BLAST query if this TRS is long enough to produce meaningful hits [9]. If the TRS is too short or unknown, there is no simple way to find the telomerase ncRNA with bioinformatics analysis alone. In this case, the combined search for all structural features occurring in the correct order in the same region of the genome could, eventually, be an alternative approach.
2.2 De novo pipeline
Contrary to the homology search pipeline, a de novo search involves looking for what we do not know, i.e., ncRNAs for which no homologous are available a priori. One of the limitations of the de novo searches is that even the most promising candidates will require experimental evidence to be validated as bona fide ncRNAs. In general, de novo ncRNA search tools [37,38] rely on the same set of assumptions: (i) the homologous sequences of a ncRNA share the same overall secondary structure; (ii) alignments of ncRNAs reveal the covariation patterns resulting from compensatory mutations and, consequently, allow the inference of the secondary structure; (iii) when applying standard folding algorithms to the alignment, the resulting minimum free energy (MFE) will be lower than the average MFE obtained by folding random sequences with the same nucleotide composition. Although none of these assumptions is always true–in particular the third one [39,40], together they can be considered as good indicators for the acceptance of a predicted candidate as a ncRNA.
In the present de novo search pipeline, we performed a whole genome Multiple Sequence Alignment (MSA) between the ten budding yeast genomes, which resulted in a set of local MSAs. The MSAs were filtered to discard too small or known protein coding regions. We then applied RNAz [37] to select the MSAs with higher probability of belonging to a ncRNA. Each selected MSA was then evaluated within its genomic context. Unannotated sequences, belonging to selected MSAs for which at least one of the sequences fall into an annotated region, are automatically given the same annotation. MSAs with no known annotation (those falling in intergenic regions for example) must be manually validated and will, eventually, represent new ncRNAs (Fig. 1B).
The automatic steps of the de novo pipeline, applied to the 10 genomes, produced 630 candidates distributed in the following way: 376 (60%) previously annotated ncRNA genes (273 of which correspond to rRNAs or tRNAs), 13 (2%) new ncRNA annotations of expected genes not found with the homology pipeline and 210 (33%) repetitive elements wrongly classified as ncRNA. The remaining 19 (3%) candidates were considered putative new ncRNA genes. Those candidates occur in intergenic regions with no previous annotations, they have a pairwise similarity higher than 50% between all sequences and display potential secondary structures supported by covariation or, at least, some compensatory mutations. The genomic location of some candidates suggests a potential regulatory role, 14 of the 19 genes occur less than 200 from the 5’ or 3’ UTR of known genes. Notice that all, except one of the candidates, were identified in only two species, a fact which prevents a more detailed analysis of the sequence variation (see Supplementary File SI 1).
Confirmation of the candidates requires experimental validation. However, the fact that 64% of the candidates could be confirmed as real ncRNA supports the assumption that, at least, some of the putative candidates correspond to real ncRNAs genes or regulatory elements. Curiously, the immediate utility of the de novo pipeline was the discovery of genes of already known families, functioning as a complement to the homology pipeline.
3 Candidate validation
Both search strategies produce many more candidates than expected. Many of the candidates (mainly those with low scores) are FPs that display some sequence or secondary structure resemblance to bona fide ncRNAs. Search tools usually assign, to each candidate, a log likelihood score that measures the ratio between the probability of obtaining the candidate using a specific model and the probability of obtaining the same candidate just by chance:
Additionally, some tools provide also an E-value for the candidate; it corresponds to the number of candidates with a score better than one would expect to obtain by chance in a sequence with the same characteristics (length and nucleotide, or di-nucleotide, composition).
Scores and E-values provide general guidance to accept or reject a candidate in a first approximation. However, they are not perfect discriminators in the sense that one cannot find a specific value of score or E-value that totally separates FP from TP candidates. In real world applications, any chosen cutoff values of score or E-value will imply a number of FP and FN. It is easy to see that the choice of cutoff value is of great importance. Choosing too high a cutoff value will discard too many positive candidates, while choosing a low value will produce a large number of FPs that will have to be manually validated one-by-one.
The final decision about each candidate must be taken over by the human curator on the basis of a combination of candidate features analysis and experience, a task often difficult or impossible to automate. To systematize the process of human validation of candidates, we established a list of acceptance criteria that must be checked: (i) Extensive sequence similarity on known conserved sequences; (ii) Candidates of families with known guide sequences (such as snoRNAs) should also present the guide sequences compatible with the targets in the same genomes; (iii) Conserved homologous synteny should be observed (similarly, known polycistronic genes should be occurring together); (iv) Known (or predicted) secondary structures should be supported by covariation and compensatory mutations in the structural alignments. The failure to comply with one or more of the above criteria would not discard a candidate per se, but it would demand stronger evidence for its acceptance.
4 Conclusions
Here we described the assembly and application of two automatic ncRNA annotation pipelines to 10 complete genomes of Saccharomycotina yeasts. Two annotation strategies were followed, a homologous search and a de novo search of ncRNAs. The assembled pipelines are based on publicly available tools and information obtained from ncRNA sequence databases. In total, we were able to find 81% of the expected ncNRAs (693 unique ncRNAs) on the searched genomes, more than doubling the 297 originally annotated ncRNAs, and 26 new candidates with no homologues in the reference species S. cerevisiae (Table 2).
Proportion of ncRNAs found with both homology and de novo pipelines, assuming the ncRNAs present in the reference genome Saccharomyces cerevisiae as 100%. Rows contain numbers of species, columns contain the percentages of ncRNA found for each family. Rows and columns legends have the same meaning as in Table 1.
| Original Annotation | Rnase P | SRP | Rnase MRP | telomerase | snRNA | snoRNA C/D | snoRNA H/ACA | Sace specific | TOTAL (count) | TOTAL | |
| sace | 86 | 1 | 1 | 1 | 1 | 5 | 44 | 29 | 4 | ||
| Fraction of found ncRNAs | |||||||||||
| cagl | 0.10 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 0.95 | 0.86 | 0.00 | 75 | 0.87 |
| zyro | 0.56 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 0.98 | 0.79 | 0.00 | 74 | 0.86 |
| sakl | 0.58 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 1.00 | 0.90 | 0.00 | 78 | 0.91 |
| klth | 0.57 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 1.00 | 0.86 | 0.00 | 77 | 0.90 |
| klla | 0.58 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 1.00 | 0.90 | 0.00 | 78 | 0.91 |
| ergo | 0.87 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 0.95 | 0.83 | 0.00 | 74 | 0.86 |
| deha | 0.09 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 0.89 | 0.66 | 0.00 | 66 | 0.77 |
| piso | 0.00 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 0.82 | 0.66 | 0.00 | 63 | 0.73 |
| yali | 0.09 | 1.00 | 0.00 | 1.00 | 0.00 | 1.00 | 0.73 | 0.45 | 0.25 | 53 | 0.62 |
| arad | 0.00 | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 0.75 | 0.48 | 0.00 | 55 | 0.64 |
| Total | 1.00 | 0.90 | 1.00 | 0.00 | 1.00 | 0.91 | 0.74 | 0.03 | 693 | 0.81 |
The analysis of the ncRNA annotation coverage, species-by-species (Fig. 5B), reveals that the less covered species are those more distant from the S. cerevisiae group. This observation shows the importance of close related species queries for homology search and suggests the need for a denser taxonomic sampling in regions of the phylogeny less represented in future genomic sequencing projects [41,42]. As an alternative hypothesis we cannot exclude that some of the ncRNAs that were not found do not exist at all. However, this hypothesis does not allow a direct bioinformatic validation. Comparing the ncRNAs found by each tool (Fig. 5A) we observe that 158 (23%) candidates are found by only one search tool, stressing the complementarity between the used methods.
Although manual validation is still needed, the human effort involved in the annotation process was strongly reduced and focused only on the validation of the automatically selected candidates and not on the search itself.
The real number of ncRNAs present in genomes is an open question [43]. In particular, data from human studies indicate that the number of potential ncRNAs could be much bigger than the currently annotated ones [44]. Although yeasts have very compact genomes (72% of the genomic sequence corresponds to protein genes) that are, on average, two hundred times smaller than the human genome, the possible existence of a number of yet unidentified ncRNAs cannot be discarded. Several recent observations such as the existence of expressed intergenic regions with no annotated function [45], the detection of several long ncRNAs of unknown function in the S. cerevisiae [16] and the extreme difficulty in identifying some elusive ncRNAs (e.g. the RNA component of the Telomerase), raise the question of how many ncRNAs are still to be found. The correct identification of possible new ncRNAs will surely require synergy between pure sequence analysis methods, high throughput techniques of sequencing [46] as well as the application of structural knowledge to ncRNA search.
5 Materials and methods
5.1 Data sources
The A. gossypii genome was obtained from the Ashbya Genome Database (agd.vital-it.ch) [23]. The A. adeninovorans genome was obtained from (C. Neuvéglise, personal communication) and the P. sorbitophila from (V. Leh, personal communication). All other genomes and the original annotations corresponding to 297 ncRNA sequences that were used as BLAST queries are from the Génolevures Database (www.genolevures.org) [22]. From the yeast genome database (www.yeastgenome.org) [47] we obtained 86 S. cerevisiae ncRNAs also used as BLAST queries. From the RFam–ncRNA families database (rfam.sanger.ac.uk) [29] (version 9.1) we downloaded 802 covariance models for INFERNAL search.
5.2 Homology pipeline BLAST search
The 383 query sequences were BLASTed against each one of the 10 genomes using the “blastall -p blastn” command (version 2.2.21) with default parameters. No query sequence was BLASTed against its own genome. The obtained candidates with E-Values < 0.1 were retained.
5.3 Homology pipeline INFERNAL search
The homology search using INFERNAL (version 1.0) was performed in two steps. First, 83 RFam covariance models, corresponding to the ncRNAs families already known in yeasts were searched and the obtained candidates with E-Values < 0.5 were retained. Second, 719 of the remaining RFam families (corresponding to all the remaining families with exception of the viral and miRNAs families) were searched and the obtained candidates with E-Values < 0.1 were retained. All INFERNAL homology searches were performed using the “cmsearch” command with default parameters.
5.4 Candidate selection
The retained candidates were automatically aligned with the known homologous fungal sequences using the “cmbuild” and “cmalign” commands from the INFERNAL package. Each alignment was manually validated according to the criteria described in the “Candidate Validation” section above.
5.5 De novo search
For the de novo search, a whole genome MSA was performed using the TBA tool [48] according to the protocol described in “A Practical Guide to Using TBA” (www.bx.psu.edu/miller_lab) with the following tree: “((((((sace cagl) zyro) (sakl klth)) (klla ergo)) (deha piso)) (yali arad))”. The MSAs smaller than 50 nts or overlapping coding regions were discarded. The remaining MSAs were split using a sliding window of 120 nts with a step of 40 nts. We searched the resulting MSAs with RNAz and retained all candidates with reported probability higher than 0.5. Retained candidates were evaluated according to sequence conservation, secondary structure prediction, possible secondary structure signals such as covariation and compensatory mutations and genomic location with respect to neighboring genes.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
The authors are very grateful to Bernard Dujon and Jean-Luc Souciet for help and numerous discussions, and for leading and animating the Génolevures Consortium. JAC is supported by the Ph.D. Program in Computational Biology of the Instituto Gulbenkian de Ciência, Portugal (sponsored by Fundação Calouste Gulbenkian, Siemens SA, and Fundação para a Ciência e Tecnologia; SFRH/BD/33528/2008).