

1 Introduction
Since 2000, the Génolevures database (URL: http://www.genolevures.org) stores sequence data and comparative results from the Génolevures Consortium's program of sequencing, annotating and analysing genomes from species in the Saccharomycotina (hemiascomycetes) subphylum, which encompasses a large evolutionary range and comprises organisms of very different physiological and ecological lifestyles. Starting in 2000 from partial genome sequence data from 13 yeast species scattered throughout this subphylum [1], in 2004 the database was greatly expanded to harbour data corresponding to four complete genomes and cross-comparisons [2], expanded again in 2008 to include those relative to complete genomes in the protoploid Saccharomycetaceae clade [3], together with external data on the two reference species Saccharomyces cerevisiae and Ashbya (Eremothecium) gossypii. Each data release was accompanied by a major update of the database and its interface [4–6].
The Génolevures database is devoted to large-scale comparisons of yeast genomes, storing not only genome sequences and genetic elements (like protein coding genes, tRNA genes, centromeres…) defined along these sequences, but also the logical relations between genes and genomes (protein families, synteny, tandem repeats…), and providing insights into topics like gene conservation, species or clade specific genes, families of proteins or chromosome shuffling, and others. The Génolevures database aims also at being a perennial repository for the data produced by the consortium, and at presenting them for consultation in a uniform format, regardless of the provenance of the data (Figs. 1 and 2).
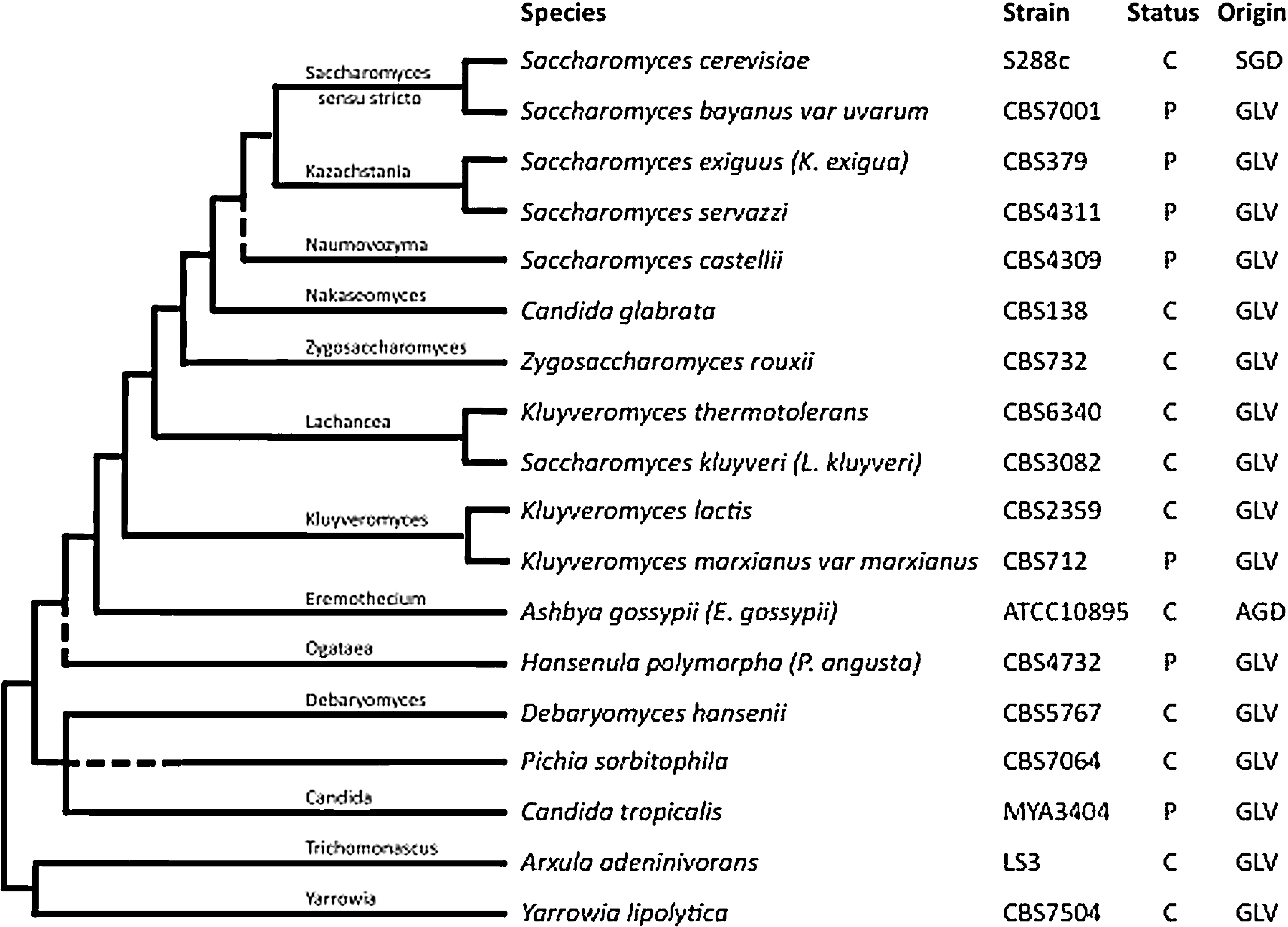
Indicative cladogram of genomes currently present in the Genolevures database, compiled from [17]. Each species is represented by a strain coming from international collections. The genome status in the database is either partial (P) or complete (C). The “Origin” column cites the databases from which the genome data are primarily stored: GLV: Génolevures online database, public or unpublished data; SGD: Saccharomyces Genome Database for the reference genome of Saccharomyces cerevisiae; AGD: Ashbya Genome Database for the reference genome of Ashbya gossypii.
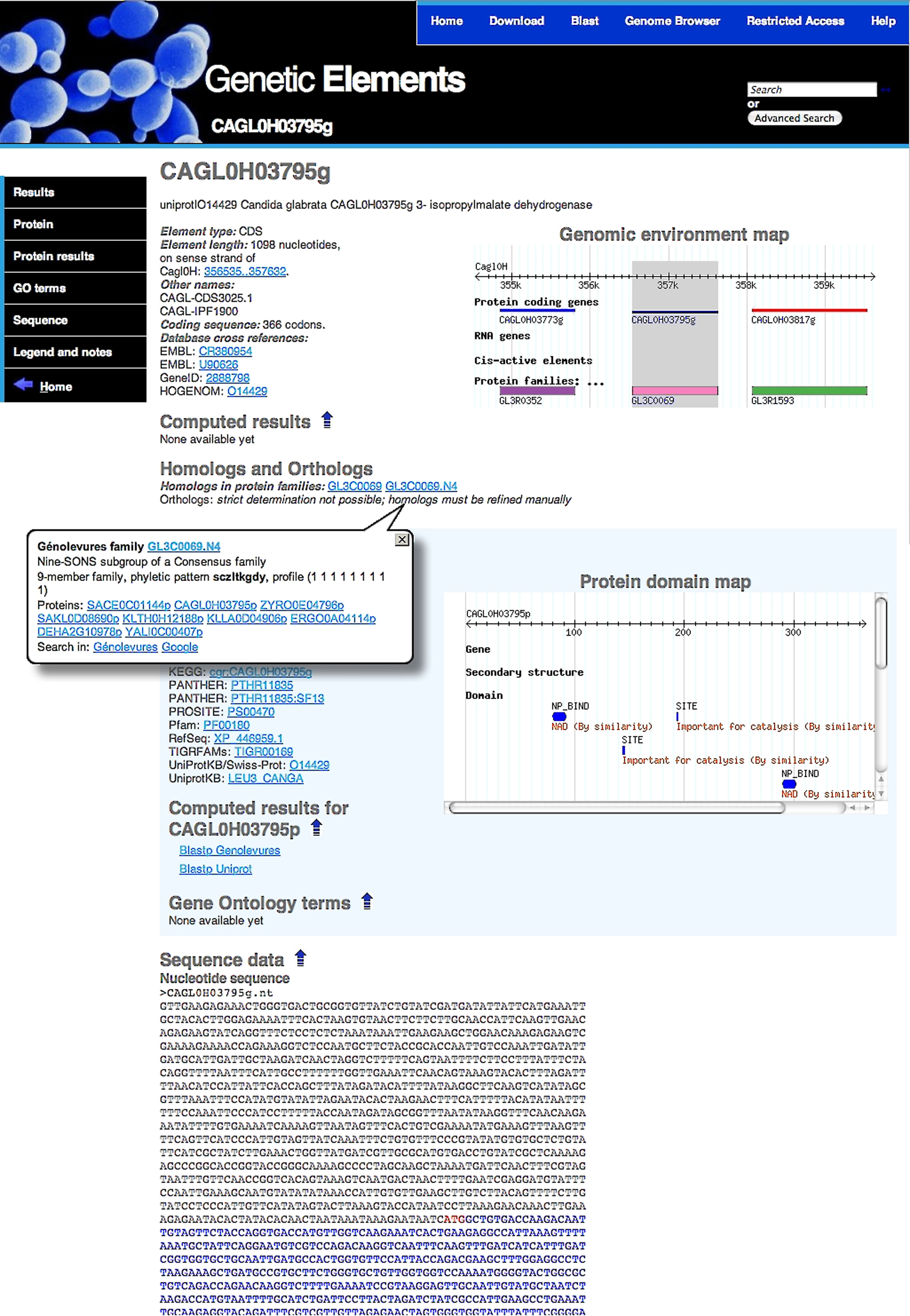
Sample Genetic Element page. The page summarizes all the available data for a given genetic element, from top to bottom: Genolevures systematic name, functional annotation, location on the chromosome, its chromosomal neighbourhood including alignment on protein family profiles (through a clickable map), membership in a protein family, domain architecture map if known, cross links to other databases, associated GO terms, nucleic acid sequence and its translation, if appropriate.
2 Implementation
The Génolevures database is designed with an object model mapped to PostgreSQL and MySQL relational databases. The use of SOFA [7] and GO [8] ontologies guarantees that the objects stored in the database are described according to widely accepted community standards. The database interface embeds the BLAST [9] tool for sequence comparisons (provided by the NCBI) and the Generic Genome Browser [10] for genome navigation (provided by the Stein lab, Cold Spring Harbor). The web interface is developed in Perl, with the BioPerl suite of modules, and the Mason web site development and delivery engine [11], the latter providing in addition an efficient page caching system.
The hardware used for the database service is composed of 8 servers (from monoprocessor to quadcore, from Pentium 4 to Xeon, from 512 Mo to 16 Go) and disk spaces (both local and in a MD1000 disk array with 10To in RAID 10) arranged in the following architecture: two host servers work together as high-availability LVS load balancers, two core and development servers are used as the support for virtual servers, two servers work as database and remote disk servers (NFS-servers), seven redundant application (HTTP and webservices) servers deployed on three real servers and four virtual servers in order to guarantee different services with minimal service interruption. All data are archived by a tape robot (8 slot LTO3 with Arkeia licence) with different periodicities according to data types.
3 Contents
From an access point of view, the database is divided into two parts: the public part where anyone can freely consult and retrieve data related to published genomes, and a private part, accessible to the consortium members, for ongoing annotation projects. Altogether, the database, sequences, annotations and computed results amount to a volume of about 3 Tbytes.
The public part currently presents data on genomes from a wide phylogenetic range, from S. cerevisiae to Yarrowia lipolytica [12]. Among the genomes of Saccharomycetaceae, the database contains genomes of species from the clades where the whole genome duplication occurred [13] such as S. cerevisiae and Candida glabrata, and species whose clades diverged before this event, like Lachancea kluyveri or L. thermotolerans.
Through the interface, users can consult a page for each genetic element which summarizes all that is stored about that element in the database: location on the chromosome, functional annotation, its chromosomal neighbourhood (through a clickable map), membership in a protein family, nucleic acid sequence and its translation if appropriate, domain architecture if known, associated GO terms and cross links to other databases. All genetic elements, whatever their kind, are named according to a unified nomenclature [14] which is unambiguous and extensible. An element name indicates the origin of the element (species, genome version and chromosome) and its relative position to other elements (elements are numbered sequentially along the chromosomes with an increment allowing insertions). For reference genomes of external origin, we also provide Génolevures-type element names after adaptation of notes and some changes in feature attributes of the elements in order to follow the Génolevures standard. In the short term, reference genomes and their annotations will remain “as is”, but in the long term, should the need arise in case of omission for example, they could be re-annotated using Génolevures procedures. The database provides, through the use of an embedded instance of the Generic Genome Browser [10], clickable and zoomable chromosomal maps showing the positions of the different kinds of elements as well as their alignments to other genomes on different tracks.
Users can also retrieve data sets computed across the different species, the most used being the protein families which reflect both expansion/contraction and universality aspects of the genes. In addition, a download page gives access to annotations and sequences in different formats. These annotations obey a structured syntax, giving the level of similarity to the reference sequence used for a given gene, the identification number of this latter sequence, its organism of origin and its common gene name together with functional annotation.
The private part holds similar data for genomes undergoing the annotation process according to the model of collaborative annotation over the Internet. Private data (up to 7 genomes, so far) are made public as soon as the annotation is completed and the main results are published. Members of the consortium can curate automatic annotations with our proprietary Magus annotation system (http://magus.gforge.inria.fr), either simultaneously on several closely related genomes or on a per genome basis, taking into account synteny relations and similarity to protein family profiles (see [6] and [15] for the building of the protein family set). The Magus genome annotation system integrates genome sequences, genetic elements placed on them, in silico analyses, and the views of external data, through an interface that only requires a Web Browser. The system allows distributed, collaborative annotation in real time, with dashboard reports that make it possible to follow the progress of the annotation community through HTML forms and graphical maps visualized through the Generic Genome Browser. Magus implements the annotation workflows elaborated for Génolevures, and obliges annotations to obey curation standards in order to guarantee the integrity and the coherency of the data. This system incorporates the widely accepted SOFA [7] and GO [8] ontologies to describe genetic elements and their annotations. Functional classification of genetic elements mainly follows that of the well-characterized species S. cerevisiae.
4 Access to data
The Génolevures database is consulted from 132 countries, according to ClusterMaps and Google Analytics, with about 37,000 visits per year (excluding search engines) made by more than 19,000 visitors (each IP address being counted as one visitor). An average consultation lasts 6 min and calls for more than 5 pages. Visitors connect to the database either directly through a bookmark in their browsers (24.4%), or by following a link from a reference site such as the Broad Institute or the NCBI (14.4%), or by using the results of a request to a search engine such as Google (61.2%).
The data stored in the database are freely accessible through a web browser (URL: http://www.genolevures.org). They are structured according to a ‘Representational State Transfer’ (REST) architecture [16], which allows users to directly build URLs for the different resources available in the database, making possible automated requests. These resources include genetic elements such as protein coding genes or ncRNA genes (/elt/species/element_name), sequences (/seq/sequence_name), protein families (/fam/family_name), chromosomal maps (/perl/gbrowse/), database search (/concordance/) and sequence comparison (/blast/) tools. For instance, for a direct access to the genetic element CAGL0D04268g, the URL is “http://www.genolevures.org/elt/CAGL/CAGL0D04268g”. In addition, a Download page allows data download in different formats (e.g. EMBL, FASTA…). A retrieval tool allowing data download after a specific query is under development.
5 Perspectives
Sequencing technology is evolving at a fast pace in terms both of quantity of data and of cost. So called Next Generation Sequencing (NGS) leads to large quantities of data comprised of sequences that are relatively small and not error free, and calls for the adaptation of the bioinformatics tools and of data processing policies. A typical project in genomics today comprises the simultaneous sequencing of several genomes from closely related species or from several strains within a single species. The increase in the number of genomes leads to a decrease in the human time available for finishing each genome, so sequenced genomes in future projects are likely to consist of a collection of contigs rather than near-complete chromosomes. In addition, the functional annotation, which up to now has been a computer-aided manual annotation in the Génolevures database, must be fully automated in the future. Leveraging the wealth of knowledge accumulated by the Génolevures consortium will make this automation possible.
The Génolevures database will continue to explore genomes of yeasts in the Saccharomycotina subphylum as well as incorporate new external reference genomes. These new genomes will be added to the database following our time-tested practices, but adapting to the new challenges from NGS data stated above. Technically, the Génolevures database can expand its scope beyond the Saccharomycotina (hemiascomycete) subphylum, and may do so to incorporate genomes belonging to other phyla within the kingdom of fungi. In addition, an increasing number of projects consist in resequencing already known genomes to obtain insights in biodiversity in terms of variant sequences. The storage of entire genomic sequences appears in this case too expensive in terms of storage space, and we will rather store differences from a reference genome, like insertions (either at the nucleotide level or at the gene level), Copy Number Variations (CNV), deletions, rearrangements and Single Nucleotide Polymorphisms (SNP). Future genome sequencing projects will likely include RNA-Seq data, which will be used by the Génolevures database to confirm gene models and their intronic structure. At this stage we consider that the integration of global RNA-Seq analyses is beyond the scope of the database, apart in the “Data sets” section and the mention on the page of a given genetic element that it is confirmed by RNA data.
In order to cope with this future influx of data and to give access to them in a reasonable time, the hardware architecture and the digital structure of data will also evolve. Indeed, the system must remain robust, expandable, and fault-tolerant, on a 24/24 7/7 basis. The foreseen increase in the amount of entries will reach a size too large for continuing to use relational databases if we want to avoid service degradation, taking into account that not only data recovery is concerned but also the use of computational applications on these data. Thus, we are taking steps to implement up-to-date computer science techniques for the evolution of the Génolevures database, such as server virtualization, grid-based web services, and distributed hash tables.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We thank all our colleagues from the Génolevures consortium for their devoted work on the sequencing, assembly and annotations of the genomes present in the database, as well as for helpful and creative discussions. The current hardware had been supported by the funding of the University of Bordeaux 1, the Aquitaine Région in the program “Génotypage et Génomique Comparée”, and the ACI IMPBIO “Génolevures En Ligne”. The upgrade of the hardware for the database is currently supported by the CNRS Institutes INS2I and INSB together with the Aquitaine Région in the project “Génolevures : outils et données en ligne pour la Génomique Comparée”. We also thank Guilhem Savel and Pascal Ung of the System and Network Administration team in LaBRI for excellent help and advice for our next system upgrade.