

1 Introduction
Plants have developed sophisticated defence mechanisms to deal with diverse unfavourable environmental factors [1,2]. Environmental stresses such as nutrient starvation, drought, high salinity, extreme temperatures and exposure to toxic chemicals can negatively impact agricultural crop yield and quality [3,4]. As an adaptive strategy, plant genomes harbour genes encoding proteins functioning as a response to stress [5–7]. Despite substantial research on responses to abiotic and biotic stresses in plants, there are still knowledge gaps regarding the molecular mechanisms underlying the diverse functions of environmental stress-associated plant genes and proteins [6]. The increasing availability of genomic sequences of members of the Viridiplantae (green algae and land plants) in conjunction with high-throughput bioinformatics tools and datasets provides new insights for examining gene families that could be central to stress responses in plants [5,7]. Genes encoding proteins with the conserved 140–160 residues USP domain (Pfam Accession: PF00582) are known to provide bacteria, archaea, fungi, protozoa, and plants with the ability to respond to different environmental stimuli [8–11]. In Escherichia coli, the Usp genes have been grouped into four classes according to their structure and amino acids sequences. They are Class I (UspA, UspC, UspD), Class II (UspF and UspG), Class III and Class IV (two USP domains of UspE) [12]. The USPA protein domain of MJ0577 (also called 1MJH) from Methanocaldococcus jannaschii crystallizes with a bound ATP, while the USPA domain of Haemophilus influenzae lacks both ATP-binding activity and residues [13]. However, the occurrence and biological significance of USPs in C. procera have not been described.
The Calotropis procera (C. procera) of the family Ascelpiadaceae is a drought-resistant, salt-tolerant wild plant species locally known as “Oshar”, which means “Giant” in English. It is an evergreen poisonous shrub naturally grown in KSA. Through its wind- and animal-dispersed seeds, it quickly becomes established as a weed along degraded roadsides, lagoon edges and in overgrazed native pastures. It has a preference for areas of abandoned cultivation, especially sandy soils with low rainfall [14,15]. It is native to west and east Africa, and south Asia, while naturalized in Australia, Central and Southern America, and the Caribbean island [15–17].
Although C. procera plant is toxic, it has many potential applications and beneficial uses. In medicine, it is both poisonous and health-giver in the same way as digitalis. The aqueous extract of C. procera (latex) inhibits cellular infiltration and affords protection against development of neoplastic changes in transgenic mouse model of hepatocellular carcinoma [18]. The root extract of C. procera has protective activity against carbon tetrachloride-induced liver damage [19]. C. procera latex is also reported to possess interesting activities such as the ability to combat diarrhoea or retard insect larval development [20,21]. Chloroform extract of roots has been reported to possess anti-inflammatory activity [22,23]. Aqueous extract of the flowers was found to exhibit analgesic, antipyretic and anti-inflammatory activity [24]. The alcoholic extracts from different parts were found to possess antimicrobial and spermicidal activities [25,26]. It has also been proven to have anti-fungal properties and can be used effectively in curing fungal diseases of the skin such as athlete's foot and ringworm [27]. Laticifer proteins (LP), recovered from the latex of this medicinal plant, are targets for DNA topoisomerase I that triggers apoptosis in cancer cell lines [28]. Also, C. procera has tannins, latex, rubber and a dye that are used in industrial practices [15]. C. procera has potentiality for bioenergy and biofuel production in semi-arid regions [29]. In ornamental field, C. procera is occasionally grown as an ornamental in dry or coastal areas because of being handsome, of a convenient size, and is easy to propagate and manage. This plant species is also recommended as a host for butterflies.
In this study, we uncovered and characterized a UspA-like gene in this medicinal plant from the de novo assembled genome contigs of the high-throughput sequencing dataset.
2 Materials and methods
2.1 Isolation of total RNA and RNAseq
RNA was the target for sequencing to avoid the presence of intron(s) in assembled sequences. Detection of exact intron sequences generally requires sophisticated problematic approaches. Three samples (reps) of leaf discs of C. procera were frozen in liquid nitrogen (approximately 50 mg tissue each) collected from upper leaves of three independent plants in the Mekka region, KSA, using Trizol (Invitrogen) and treated with RNase-free DNase (Promega). Sampling was done in the midday, as the plants are under heat and drought stresses that might help in the recovery of high-coverage of Usp-responsive transcripts. RNA samples were sent to Beijing Genomics Institute (BGI), Shenzhen, China for deep sequencing and datasets were provided for de novo assembly and bioinformatic analysis.
2.2 Sequence de novo assembly
The raw sequence data were obtained using the Illumina python pipeline v. 1.3. For obtained libraries, only high-quality reads (>20) were retained. Then, de novo assembly of the obtained short pair-end read datasets was performed using assembler Velvet [30] followed by creation of putative unique transcript (PUTs) with a combination of different k-mer lengths and expected coverage. In total, the yielded expressed sequence tag (EST) assemblies were merged into Solanum lycopersicum cDNA, clone: LEFL1037AD03, HTC in leaf (accession No. AK322367.1), where the identity of sequences was over 95% and 40 bp overlapping. This clone is identified as a Usp-like cDNA sequence in which the USP domain is located between bases 200 and 650 [31].
2.3 Basic local alignment search tool (BLAST)
BLAST was used to find regions of local similarity among Usp related sequences. The program was used to compare the recovered nucleotide and deduced amino acids sequences to sequence in the NCBI database, and calculates the statistical significance of matches based on pair-wise alignment method. BLAST was also used to infer functional and evolutionary relationships among the resulted sequences as well as help identifying members of UspA-like gene families (http://www.ncbi.nlm.nih.gov/BLAST).
2.4 AlignX and ClastalW
CLUSTAL [32] is a general purpose multiple sequence alignment program for DNA, RNA or protein. It produces biologically meaningful multiple sequence alignments of divergent sequences and calculates the best match for the selected sequences. Then, it aligns them up so that the identities, similarities and differences can be distinguished. Evolutionary relationships can be viewed via Cladograms or Phylograms. AlignX® Module (rapid multiple sequence alignment with minimal preparation AlignX®) uses a modified CLUSTAL algorithm to generate multiple sequence alignments of either protein or nucleic acid sequences for similarity comparisons and for annotation. The power of AlignX® is in maintaining annotated features within the alignment for easy visualization and localization of regions of interest.
2.5 Determination of phylogenetic relationships
The unweighted pair group method with arithmetic mean (UPGMA) method was used to build a tree where the evolutionary rates are free to differ in different lineages. To evaluate the reliability of the inferred trees, CLC Genomics Workbench (version 3.0) was used to allow the option of doing a bootstrap analysis. A bootstrap value is attached to each branch as a measure of confidence. The GenBank accession numbers for Usp cDNA sequences utilized in this work are shown in Table 1.
Accession numbers, description of the genes and organisms with highest similarities to C. procera UspA-like cDNA sequence (accession No. KC954274).
| Accession No. | Description | Organism (Latin name) |
| NP_566564 | Universal stress protein (USP) family | Arabidopsis thaliana |
| NP_566991 | Universal stress protein (USP) family | Arabidopsis thaliana |
| XP_002271154 | PREDICTED: universal stress A-like protein | Vitis vinifera |
| XP_002275745 | PREDICTED: universal stress A-like protein | Vitis vinifera |
| XP_002277653 | PREDICTED: universal stress A-like protein isoform 1 | Vitis vinifera |
| XP_002281607 | PREDICTED: universal stress A-like protein | Vitis vinifera |
| XP_002316036 | Predicted protein | Populus trichocarpa |
| XP_002519217 | Conserved hypothetical protein | Ricinus communis |
| XP_002877951 | Universal stress protein family | Arabidopsis lyrata subsp. Lyrata |
| XP_002885183 | Universal stress protein family | Arabidopsis lyrata subsp. Lyrata |
| XP_003529116 | PREDICTED: universal stress A-like protein | Glycine max |
| XP_003547361 | PREDICTED: universal stress A-like protein | Glycine max |
| XP_003567271 | PREDICTED: universal stress A-like protein | Brachypodium distachyon |
| XP_003568332 | PREDICTED: universal stress A-like protein | Brachypodium distachyon |
| XP_003568882 | PREDICTED: universal stress A-like protein | Brachypodium distachyon |
| XP_003569311 | PREDICTED: universal stress A-like protein | Brachypodium distachyon |
| XP_003594749 | Universal stress A-like protein | Medicago truncatula |
| XP_003616191 | Universal stress A-like protein | Medicago truncatula |
2.6 The 3D homology modelling
The 3D homology protein modelling was carried out using Swiss-Model, a protein-modelling server, accessible via the EXPASY (http://www.expasy.org/). Superimposition of C. procera USPA-like predicted protein model on Thermus thermophilus USP UniProt accession No. Q5SJV7 (http://www.uniprot.org/) was constructed using RasMol (http://www.umass.edu/microbio/rasmol/) and Deep-View program (http://spdbv.vital-it.ch/). The functional domains were identified from the NCBI conserved domain database (CDD) (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml/) and UniProtKB protein knowledge base (http://www.uniprot.org/). Software uses 3D structure information to explicitly define domain boundaries and provide insights into sequence structure/function relationships, as well as domain models imported from a number of external source databases (Pfam, SMART, COG, PRK, TIGRFAM).
2.7 Structure alignment
DaliLite was used as the structural alignment method on representative datasets [34–36]. Protein 3D structure comparison depends on the alignment algorithm, the similarity measure, and the fractions of the protein structures considered for the pairwise structure alignment [33]. Model of C. procera USPA-like deduced amino acids sequences was built on the Thermus thermophilus USP UniProt accession No. Q5SJV7. The protein model and its 3D structure were used in pairwise comparison of protein structures using DaliLite program server at EBI (http://www.ebi.ac.uk/Tools/dalilite/) [37] and protein alignment was done to identify conserved and diverse structure domains. Root mean square deviation (RMSD), measuring the average distance between the backbone of superimposed proteins, was detected using DaliLite according to the following formula:
3 Results
3.1 Nuclear genome sequence (NGS)
Nuclear genome paired-end short-sequence reads of C. procera cDNAs were generated using the Illumina Genome Analyser IIx (GAIIx) according to manufacturer's instructions (Illumina, San Diego, CA). Assemblies were mapped to the available Arabidopsis thaliana Usp sequence (accession No. NP_112578.4). The resulted C. procera contigs were blasted and the best hit (Vitis vinifera, accession No. XP_002277653.1) was used in a second iteration of mapping. BLAST analysis for the resulted contig was done and a third iteration of mapping was done against the next best hit (Catharanthus roseus accession No. XFD420803) using SAOP [38]. The number of reads aligned was 17 551, with an average coverage of ∼2933. The length of consensus sequence equals 542 nt. ORF analysis showed a full-length ORF with a start codon at bases 48–46 and a stop codon at bases 537–539 (Fig. 1a). The recovered UspA-like cDNA and deduced amino acids sequences were deposited in the NCBI and received the accession Nos. KC954274 and AGT02387, respectively.
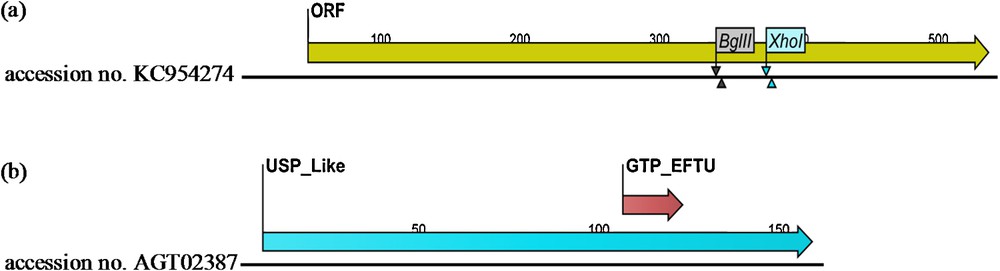
(Colour online) a: ORF restriction map for the obtained Usp-like cDNA sequence (accession No. KC954274). This sequence was characterized by the presence of BglII site (at 340-345 nt) and XhoI (at 376-381 nt); b: USP and GTP-EFTU domains of the deduced amino acids sequence (accession No. AGT02387) (with a length of 489 aa) of the obtained USP-like deduced amino acids sequence as analyzed by Pfam database.
The deduced amino acids sequence (with a length of 489) obtained from ORF analysis was analyzed against the Pfam database conserved domain database and UniProtKB protein knowledge base to locate protein domains. Domain analysis revealed the presence of a universal stress protein domain (conserved domain database accession No. CD00293 and Pfam database accession No. PF00582) (Fig. 1b). Function of this domain was evolutionarily conserved in numerous prokaryotic as well as eukaryotic organisms [9,39]. Generally, Usp genes play an active role in the abiotic stress response, but their function in plant remain largely unknown [40]. Conserved domain analysis also revealed a second domain belonging to elongation factor Tu GTP binding domain (Pfam database accession No. PF00009). The GTP–EFTU binding domain (Fig. 1b) is involved in a conformational change mediated by the hydrolysis of GTP to GDP [41].
3.2 BLAST analysis
BLAST (either protein-protein BLAST or BLASTp) was performed to identify sequence similarity with homologous USP-like amino acids sequences from other organisms. The interpretation of the score and sequence similarity from BLAST searching eventually led to the identification of putative homologous protein sequences. Results for the most closely-related amino acids sequence to C. procera USP-like predicted protein indicated that the PREDICTED: USPA-isoform 1 of Vitis vinifera has the lowest e-value (1 × e−87). These results indicate that the speculated C. procera USP can be a member of USPA subfamily (Table 2).
Accession numbers, description (organism latin name) and the calculated e-value of homologous proteins to C. procera USP-like deduced amino acids sequence (accession No. AGT02387) identified using BLASTp search.
| Accession | Description | Total score | Query cover (%) | E value | Max identity (%) |
| XP_002277653.1 | PREDICTED: universal stress A-like protein isoform 1 [Vitis vinifera] | 264 | 99 | 1.00E-87 | 79 |
| XP_002519217.1 | Conserved hypothetical protein [Ricinus communis] | 263 | 100 | 2.00E-87 | 78 |
| XP_002316036.1 | Predicted protein [Populus trichocarpa] | 261 | 99 | 7.00E-87 | 75 |
| XP_002885183.1 | Universal stress protein family [Arabidopsis lyrata subsp. lyrata] | 238 | 98 | 1.00E-77 | 72 |
| NP_566564.1 | Universal stress protein family [Arabidopsis thaliana] | 236 | 98 | 7.00E-77 | 71 |
| XP_003547361.1 | PREDICTED: universal stress A-like protein [Glycine max] | 233 | 98 | 8.00E-76 | 70 |
| XP_003568882.1 | PREDICTED: universal stress A-like protein [Brachypodium distachyon] | 215 | 98 | 2.00E-68 | 64 |
| XP_003594749.1 | Universal stress A-like protein [Medicago truncatula] | 207 | 98 | 1.00E-65 | 63 |
| XP_003569311.1 | PREDICTED: universal stress A-like protein [Brachypodium distachyon] | 196 | 100 | 2.00E-61 | 59 |
| XP_003568332.1 | PREDICTED: universal stress A-like protein [Brachypodium distachyon] | 190 | 98 | 1.00E-58 | 59 |
| XP_002275745.1 | PREDICTED: universal stress A-like protein [Vitis vinifera] | 184 | 98 | 2.00E-56 | 56 |
| XP_002271154.1 | PREDICTED: universal stress A-like protein [Vitis vinifera] | 184 | 98 | 2.00E-56 | 59 |
| XP_003567271.1 | PREDICTED: universal stress A-like protein [Brachypodium distachyon] | 180 | 98 | 6.00E-55 | 55 |
| XP_002281607.1 | PREDICTED: universal stress A-like protein [Vitis vinifera] | 178 | 98 | 3.00E-54 | 56 |
| XP_003529116.1 | PREDICTED: universal stress A-like protein [Glycine max] | 172 | 98 | 8.00E-52 | 53 |
| XP_002877951.1 | Universal stress protein family [Arabidopsis lyrata subsp. lyrata] | 171 | 98 | 2.00E-51 | 54 |
| NP_566991.2 | Universal stress protein (USP) family [Arabidopsis thaliana] | 171 | 100 | 2.00E-51 | 53 |
| XP_003616191.1 | Universal stress A-like protein [Medicago truncatula] | 171 | 98 | 2.00E-51 | 52 |
3.3 Multi-sequence alignment (MSA) and phylogenetic analysis
The best BLAST search hits were used to perform multi-sequence alignment. This resulted in 18 sequences originating from seven different plant species. Alignment of these sequences to the newly recovered sequence (accession No. KC954274) was obtained by a gap open penalty of 10 and a gap extension penalty of one. Sequences with more than 50% identity with the obtained C. procera USPA-like deduced amino acids sequence were used (Table 3 and Fig. 2). The results also showed that the closest sequence to that of C. procera USPA-like deduced amino acids sequence is that of Vitis vinifera PREDICTED: universal stress A-like protein isoform 1 (accession No. XP_002277653.1). These results support the obtained BLAST results. MSA results were used to perform phylogenetic tree for the 19 proteins and results (Fig. 3) were similar to those of previous analyses.
Pairwise alignment between each USP hit sequences as compared to the obtained sequence of C. procera USPA-like deduced amino acids sequence (accession No. AGT02387).
| Accession No. | Gaps | Differences | Differences | Identity % | Identities |
| XP_002277653.1 | 7 | 47 | 0.33 | 71.86 | 120 |
| XP_002519217.1 | 6 | 46 | 0.33 | 72.29 | 120 |
| XP_002316036.1 | 6 | 54 | 0.4 | 67.47 | 112 |
| XP_002885183.1 | 1 | 53 | 0.4 | 67.48 | 110 |
| NP_566564.1 | 1 | 53 | 0.4 | 67.48 | 110 |
| XP_003547361.1 | 6 | 56 | 0.42 | 66.27 | 110 |
| XP_003594749.1 | 13 | 67 | 0.53 | 59.64 | 99 |
| XP_003569311.1 | 4 | 73 | 0.61 | 55.21 | 90 |
| XP_003568882.1 | 9 | 73 | 0.57 | 57.31 | 98 |
| XP_002877951.1 | 8 | 79 | 0.66 | 52.41 | 87 |
| NP_566991.2 | 6 | 79 | 0.67 | 51.83 | 85 |
| XP_002275745.1 | 10 | 81 | 0.67 | 51.79 | 87 |
| XP_002281607.1 | 10 | 89 | 0.77 | 47.02 | 79 |
| XP_003529116.1 | 5 | 79 | 0.68 | 51.23 | 83 |
| XP_003568332.1 | 7 | 77 | 0.63 | 54.17 | 91 |
| XP_003567271.1 | 10 | 82 | 0.69 | 51.19 | 86 |
| XP_002271154.1 | 8 | 81 | 0.68 | 51.5 | 86 |
| XP_003616191.1 | 10 | 86 | 0.73 | 49.11 | 83 |
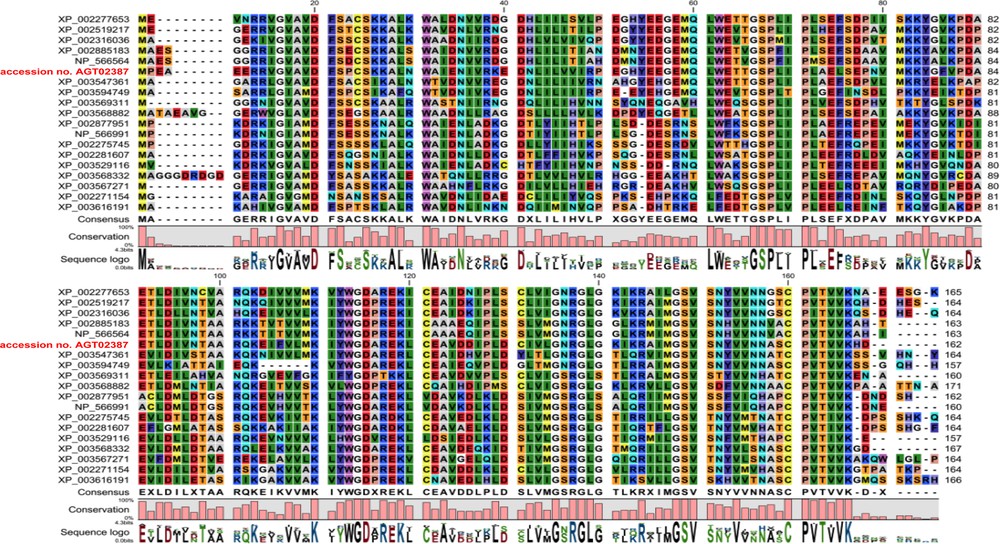
(Colour online) Multi-sequence alignment of the 18 USP sequences with the obtained C. procera USPA-like deduced amino acids sequence (accession No. AGT02387).
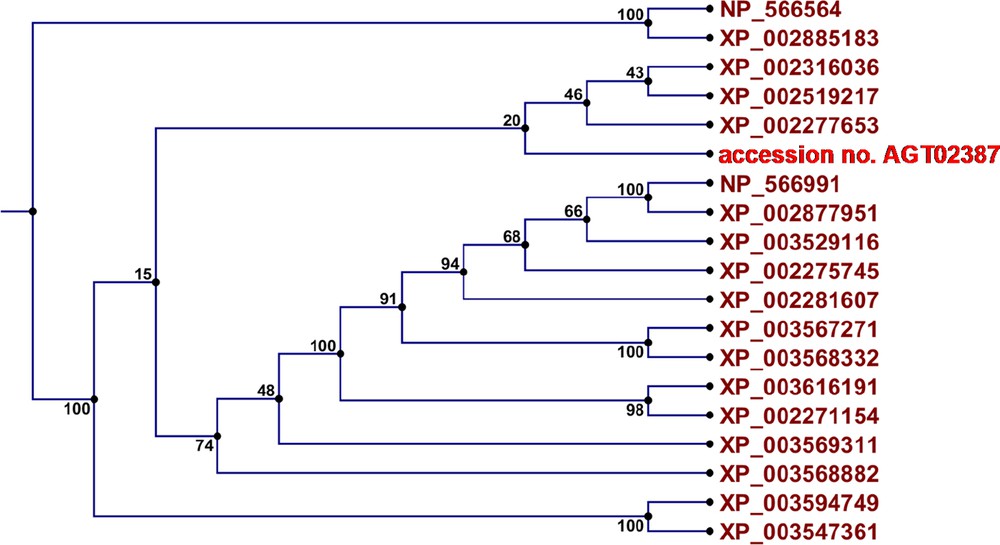
(Colour online) Phylogenetic analysis of 18 USP proteins and C. procera USPA-like deduced amino acids sequence (accession No. AGT02387).
3.4 3D structure modelling
USPs exist as homodimers, and genetic studies showed that their cellular assignments are extensive, including functions relating to stress resistance, carbon metabolism, cellular adhesion, motility, and bacterial virulence. Some recent articles suggest that USPs present as heterodimers [42]. Using multi-sequence alignment, we were able to identify important functional domains and motifs within C. procera USPA-like predicted protein sequence (Fig. 4a, b). USP proteins are characterized by the presence of ATP/AMP-binding motif residues indicating that they may be regulated by ATP. Ten ligand-binding sites were predicted based on multi-sequence alignment to the ATP-binding universal stress protein [43]. Table 4 shows a list of predicted proteins that bind to AMP, along with their position and binding sites. Based on structural alignment, a theoretical 3D model for C. procera USPA-like deduced amino acids sequence was created (Fig. 5), corresponding to residues 6-160 of the primary structure. The predicted model was created using the Swiss-Model, protein-modelling server. The overall dimensions of the predicted model are 58.547 Å × 45.719 Å × 30.844 Å.
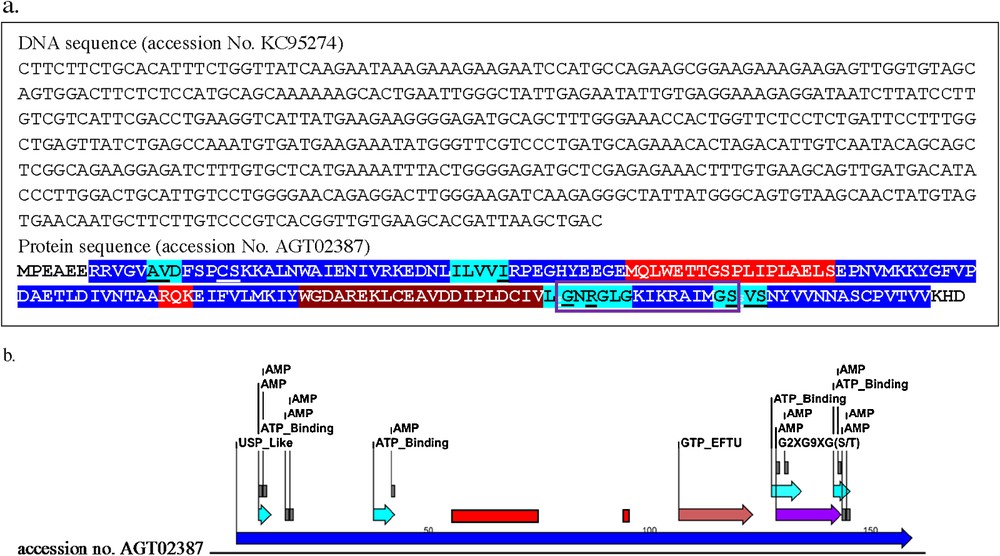
(Colour online) Identified cDNA sequence (accession No. KC95274): a: and protein (accession No. AGT02387) motifs (a,b) in the obtained Calotropis USPA-like cDNA and deduced amino acids sequences. USP domain = blue, ATP binding domain = turquoise, ATP/AMP binding residues = underlined, GTP-EFTUD domain = brown, G2XG9XG(S/T) domain = violet box, regions with no superimposition to other closely related USP 3D models = red.
Predicted deduced amino acids of the C. procera USPA protein (accession No. AGT02387) that binds to AMP along with their positions and binding sites.
| Amino acid | Position | Binding site |
| Ala | 12 | AMP, via carbonyl oxygen |
| Val | 13 | AMP, via carbonyl oxygen |
| Cys | 18 | AMP, via carbonyl oxygen |
| Ser | 19 | AMP |
| Val | 41 | AMP, via amide nitrogen and carbonyl oxygen |
| Gly | 129 | AMP, via amide nitrogen and carbonyl oxygen |
| Arg | 131 | AMP |
| Ser | 143 | AMP |
| Val | 144 | AMP, via amide nitrogen |
| Ser | 145 | AMP |
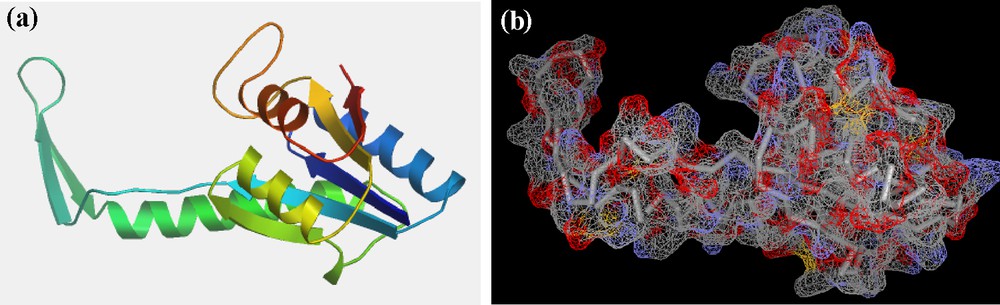
(Colour online) Theoretical 3D model for C. procera USPA-like deduced amino acids sequence (accession No. AGT02387): a: α-helix, β-sheets and turns; b: calculated surface of Calotropis USPA-like deduced amino acids sequence.
Multi-sequence alignment also showed that residues making contacts with ATP/AMP exist in five domains. The first domain (Ala (A), Val (V) and Asp (D)) is involved in coordinating a Mn2+ ion, which in turn binds to the phosphate groups. This residue is conserved in most of the bacterial and Arabidopsis sequences. The Ile (I) at position 41 of the second domain (Ile (I), Leu (L), Val (V), Val (V) and Ile (I)) binds to adenine with a hydrogen bond. This position is conserved in many of the aligned sequences. The G residue at position 129 of the third domain (Leu (L), Gly (G), ASN (N), Arg (R), Gly (G), Leu (L) and Gly (G)) binds to ribose with a hydrogen bond. This performance exists in nearly all the USPA sequences. Position Arg (R) at position 130 of the same domain binds with a hydrogen bond at the beta phosphate group, which is conserved in all aligned USPA sequences. Ser (S) at position 143 of the fourth domain (Gly (G), Ser (S), Val (V), and Ser (S)) binds to the gamma phosphate group with a hydrogen bond, which is conserved in all the aligned USPA sequences. Also, Ser (S) at position 145 of the same domain binds to the alpha phosphate with a hydrogen bond. This residue is also conserved in all aligned USPA sequences [44]. The G2XG9XG(S/T) domain is also identified in C. procera USPA-like deduced amino acids sequence from bases 129 to 143. All Identified residues and domains are shown in Fig. 4a, b.
3.5 Structure alignment
We used DaliLite to superimpose C. procera USPA-like model on Thermus thermophiles USP protein 3D structures (protein database accession No. 2Z3 V), which was downloaded from protein database. Accession No. 2Z3V is the closest homologous protein sequence with the obtained C. procera USP 3D predicted structure (Fig. 5). To proof the accuracy of our theoretical 3D model of C. procera USPA-like deduced amino acids sequence, we used DaliLite to compute optimal and suboptimal structural alignments between T. thermophiles USP protein and the theoretical 3D model of C. procera USPA-like amino acids sequence. The resulting superimposition is shown in Fig. 6 with Z-score of 26.7, and a number of equivalent residues of 135 and RMSD of 0.3. The figure also shows that 3D model of C. procera USPA deduced amino acids (yellow) has almost the same coordinates of T. thermophiles USP protein (grey) except at two positions, i.e., A (residues 53-70) and B (residues 95–97), which are hyper-variant regions. Regions A and B are located upstream the GTP-EFTU domain in an area that is not involved in any important identified domain. These results support our finding of obtaining a C. procera sequence belonging to USPA subfamily. Also, the results proved the accuracy of our theoretical 3D modelling for C. procera USPA-like deduced amino acids sequence.
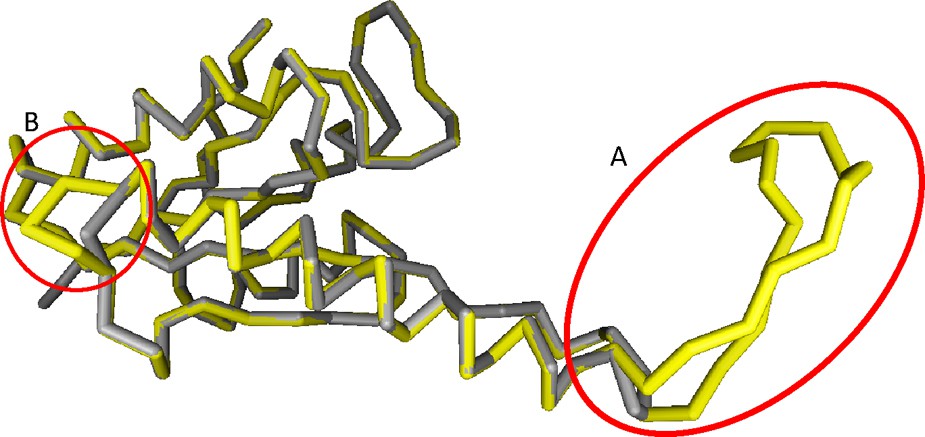
(Colour online) Superimposition between T. thermophiles USP amino acids sequence and the C. procera USPA deduced amino acids sequence (accession No. AGT02387). A & B shows first and second hyper-variant regions. Superimposition of C. procera USPA-like model was constructed on Thermus thermophilus USP UniProt accession No. Q5SJV7 (http://www.uniprot.org/) using RasMol (http://www.umass.edu/microbio/rasmol/) and Deep-View program (http://spdbv.vital-it.ch/).
4 Discussion
The obtained C. procera sequence showed features for a universal stress protein class A (USPA) domain. USPA protein domain whose features determine its function has been evolutionarily conserved in numerous prokaryotic as well as eukaryotic organisms [9,39]. Proteins with the USP domain are known to provide plants with the ability to overexpress and respond to environmental stresses such as nutrient starvation, drought, high salinity, extreme temperatures and exposure to toxic chemicals [45]. These conditions are normal for a desert wild plant, like C. procera. Conserved domain analysis revealed a second domain belonging to elongation factor Tu GTP binding domain (Pfam database accession No. PF00009) [41]. Some evidence showed that a number of EF-Tu protein group members are interacting with some proteins belongs to USP protein family in E. coli [46]. This is the first evidence to show that some USP proteins have EF-Tu activity.
To assign the identified USP protein to its appropriate protein subfamily, several tests were conducted. First, we used BLAST search against Genbank protein database. The interpretation of the score and sequence similarity led to the identification of putative homologous protein sequences. Results showed that the most closely-related gene was USPA-isoform 1 of Vitis vinifera, which showed the lowest e-value (1 × e−87). These results indicate that the speculated C. procera USP protein can be a member of USPA subfamily. The best BLAST search hits were used to perform multi-sequence alignment and 18 sequences were resulted. Alignment of the 19 sequences (Table 3 and Fig. 2) showed that the closest sequence to the obtained C. procera USPA-like amino acids sequence is Vitis vinifera PREDICTED: universal stress A-like protein isoform 1 of accession No. XP_002277653.1. This result also supports the hypothesis that C. procera USP can be a member of USPA subfamily. MSA results were used to perform phylogenetic tree for the 19 USP-like proteins and results (Fig. 3) were similar to those of previous analyses. Multi-sequence alignment also proved that all important functional domains and motifs belong to USP protein family are located within C. procera USPA-like deduced amino acids sequence (Fig. 4a,b). USPs show the presence of ATP/AMP-binding motif residues indicating that they may be regulated by ATP. Table 4 shows a list of predicted amino acids that bind to AMP, along with their positions and binding sites. Multi-sequence alignment also showed that residues making contact with ATP/AMP exist in five domains within C. procera USP-like sequence. These residues are conserved in all aligned sequences [44].
Finally, the 3D Superimposition analysis between T. thermophiles USP amino acids sequence and the constructed C. procera USPA predicted protein has almost the same coordinates except in two positions, i.e., A (residues 53-70) and B (residues 95-97), which are hyper-variant positions. Regions A and B are located upstream the GTP-EFTU domain in an area that is not involved in any important identified domain. These results support our finding of obtaining a C. procera protein sequences belonging to USPA subfamily. Also, the results prove the accuracy of our theoretical 3D modelling for the obtained C. procera USPA-like deduced amino acids sequence. Further study to detect the regulation of this gene under abiotic stress conditions is underway. The gene will also be overexpressed in a model transgenic plant, like tobacco or Arabidopsis, through transgenesis in order to detect the efficacy of introducing this gene in conferring abiotic stress tolerance in plant.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant No. 1432-3-15/HiCi. The authors, therefore, acknowledge with thanks DSR technical and financial support.