

1 Introduction
Metallothioneins (MT) are well conserved ubiquitous metal-binding proteins that constitute a large superfamily of small, cysteine-rich proteins involved in homeostasis of essential metals and in protecting cells against heavy-metal toxicity. From its first discovery in the cadmium-treated horse kidney [1] and [2], a multitude of MT genes and proteins have been identified in a wide variety of organisms [3]. A search on MT genes in the EMBL Nucleotide Sequence Database retrieved about 2330 MT mRNAs, half of them being represented by genes from vertebrates and, between the latter, about 75% are from mammals, followed by the MT genes from fishes (more than 200 mRNAs); few MT genes have been described in aves, reptiles, and amphibians.
In mammals, four tandemly clustered genes (MT1 to MT4) have been identified [4]; all genes encode for conserved peptide chains retaining 20 invariant metal-binding cysteines; MT3 and MT4 have developed, however, additional properties relatively to MT1 and MT2, such as protection against brain injuries [5–7] and epithelial differentiation [8], respectively. In addition, during the evolution of the lineage that led to modern humans, MT1 has undergone further duplication events that have resulted in 13 duplicate isoforms (MT1a to MT1m) [9]. From them, the human metallothionein Ib (hMT1b) shows the presence of an additional cysteine, forming a Cys–Cys–Cys triplet at the level of the C-terminus β-domain [10]. Interestingly, a high level of expression of the endogenous hMT1b gene has been detected only in human hepatoma and renal carcinoma cell lines [10].
In the other vertebrates, so far only one or two distinct isoforms have been identified [11]. In the last past years, however, more MT sequences from the “neglected” tetrapods have been identified and many articles that shed new light on the evolution of vertebrates MTs have been published [9,11–15]. In addition, the increasing availability of annotated genomic databases has provided a further resource to identify new genes and to study their functional diversification and evolution [16,17]. To date, in amphibians a single MT transcript has been cloned and sequenced [13,15]; in the African clawed frog Xenopus laevis, the MT isoform so far investigated, termed MTA [18], is made of 62 amino acids with 20 cysteines.
Exploiting the annotation of the Silurana tropicalis genome, we performed a search for MT-like sequences. Such analysis led to the identification of a new MT gene in the western clawed frog genome, exhibiting all the features to be considered an active coding region. However, surprisingly enough, this new isoform shows unexpected diversity in the primary sequence, such as the presence of 17 cysteine residues instead of the canonical 20 cysteines of vertebrate MTs, as well as the clustering of some of these cysteine residues into Cys–Cys–Cys sequences instead of the Cys–X–Cys, Cys–Cys and Cys–X–Y–Cys sequences typical of the 20 cysteine residues of vertebrate MTs. A further search in the database of Expressed Sequence Tags (dbEST) at the NCBI portal allowed us to identify a cDNA fragment (da89f03.y1) from the X. laevis tadpole stage 24 EST (LIBEST_004113) library, whose sequence corresponds to the MT identified on the western clawed frog genome.
In the light of these bioinformatics data, we decided to verify the expression of this atypical MT isoform in two different larval phases, stages 24 and 48 respectively [19], and in two adult tissues of the anuran X. laevis. In particular, we chose the skin and the liver: the first in amphibians acts as a barrier and respiratory organ, the latter is a detoxifying organ and the main site of MT expression and synthesis. Analyses of Real-Time PCR demonstrate the constitutive expression of this atypical MT both in tadpoles and adult tissues, with the highest amount of transcripts in liver. Phylogenetic analyses demonstrate that this uncommon Xenopus MT sequence is the most derived among amphibian MTs; Tajima's relative rate test also demonstrates in X. laevis a different evolutionary rate between the canonical MTA and this novel MT isoform herein described.
2 Materials and methods
2.1 Animals
Adult X. laevis were obtained from Nasco (Fort Atkinsons, Wisconsin, USA). They were kept and used at the Department of Biology of the University of Naples, Federico II, according to the guidelines and policies dictated by the University Animal Welfare Office. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of Naples Federico II (Permit Number: 2014/0017970). All procedures were performed according to the Italian Ministry of Health (Authorization DL 116/92) and European regulations on the protection of animals employed for experimental and other scientific purposes. All surgery was performed under ethyl 3-aminobenzoate methanesulfonate (MS-222, Sigma); animals were killed by decapitation. All trials were adopted to minimize suffering. To obtain eggs, X. laevis females were injected in the dorsal lymphatic sac with 500 units of Gonase (AMSA) in amphibian Ringer solution (111 mM NaCl, 1.3 mM CaCl2, 2 mM KCl, 0.8 mM MgSO4, in 25 mM Hepes, pH 7.8). Fertilized eggs and embryos were obtained by standard insemination methods [20] and staged according to Nieuwkoop and Faber [19].
2.2 Bioinformatics methods for the MTs analysis
The nucleotide primary structures of the two X. laevis MTs (Q05890 and da89f03.y1) were aligned using the Clustal Omega tool. Owing to the high sequence similarity shared by the two different MTs, specific primers for conventional and Real-Time PCR analyses were designed using as a template the most divergent regions of the MT identified as an EST result in the X. laevis tadpoles (da89f03.y1).
2.3 Collecting total RNA from embryonic and adult clawed frog tissues
X. laevis tadpoles bred in 1/10 Ringer were collected at stages 24 and 48. For each stage, three tadpoles were pooled in three different groups and stored in RNA-later at –20 °C. Liver and skin tissues from three different adults were dissected and stored individually in RNA-later at –20 °C. Samples were homogenized and suspended in TriZol (Life Technologies) by repipetting, and then total RNAs were extracted into the aqueous phase, precipitated with isopropanol, washed with 75% ethanol and then resuspended into nuclease free water.
2.4 RNA purification and cDNA synthesis
The quality of each total RNA was checked by electrophoresis on 2% agarose gel stained with ethidium bromide and measuring the optical density at 260/280 nm. A ratio of 1.8–2.0 was accepted for further reverse transcription. QuantiTect Reverse Transcription Kit (Qiagen) was used for the removal of genomic DNA contamination and for subsequent cDNA synthesis. Approximately 1 μg of total RNA was used, according to the kit's protocol.
2.5 Cloning and sequencing
The cDNA corresponding to a fragment of the new MT isoform was cloned starting from reverse-transcribed RNA of stage 24 tadpoles. Conventional PCR reactions were carried out on 2 μL of first-strand cDNA, using the following two MT specific primers designed as described above: forward, 5’-CCCTGCTGCTGTAAAGGCG-3’; reverse, 5’-GTGCAAAGAAGATCACTGGGG-3’. The purified PCR product was directly cloned into the pSC-A vector using the StrataClone PCR Cloning Kit (Agilent Technologies) according to the manufacturer's instructions. Several randomly selected colonies were inoculated into LB broth containing 100 μg/mL ampicillin and incubated overnight at 37 °C while shaking. Plasmids containing inserted sequences were purified using Fast Plasmid Mini kit (5 Prime) and sequenced using automated methods on an ABI PRISM Genetic Analyzer (PE Biosystems). The sequence of this MT cDNA fragment appears in the DDBJ/EMBL/GenBank database with accession number LN681256.
2.6 Real-time RT-PCR analysis
Target gene mRNA was quantified with quantitative Real-Time PCR. All analyses were carried out using the Applied Biosystem 7500 Real-Time PCR System and the Power SYBR® Green PCR Master Mix (Life Technologies) following procedures recommended by the manufacturer. The amplification was set at 20 μL; each reaction containing 12 μL of real-time PCR Master Mix, 1 μL of each of the forward and reverse primer (10 μM), 2 μL of cDNA diluted 1:1 and 4 μL of nuclease free water. Nuclease-free water for the replacement of cDNA template was used as a negative control. Reaction was carried out with an initial step at 1 min at 95 °C, followed by 40 cycles of 95 °C for 15 s at and 60 °C for 40 s. A melting curve analysis of PCR products was performed from 60 to 95 °C in order to ensure gene-specific amplification. All the data are expressed relative to Histone-H4, which was used as a housekeeping reference to normalize gene expression levels in each sample [20]. Changes in the gene expression relative to the different samples were calculated according to the standard 2−ΔΔCt method described by Livak and Schmittgen [21]. MT primers used were described above, the H4 primers were: forward, 5’-CGGGATAACATTCAGGGTA-3’; reverse, 5’-TCCATGGCGGTAACTGTC-3’.
2.7 Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) from three separate experiments in each sample. Statistical analyses were carried out by StatView software (Altura Software, Inc.). Differences between mean values were analysed by one-way analysis of variance (ANOVA) followed by Fisher's LSD test. The differences were considered significant when P < 0.05.
2.8 Multiple alignments of sequences and phylogenetic analyses
Sequences of vertebrate MTs used for the phylogenetic analyses were retrieved from the NCBI database. The multiple alignment of MT amino acid sequences was obtained using program MUSCLE version 3.8 [22] and refined manually by program Se-Al v. 20a11. The accession numbers of the selected sequences and the alignment are available upon request.
Phylogenetic trees were inferred by the Maximum Likelihood (ML) model. Analyses were performed with SeaView version 4 software [23], using the Dayhoff + I + G (Invariant sites and Gamma-distributed rates for sites) substitution model following the results from ProtTest [24]. Bootstrap branch support was estimated using 1000 data sets. Tree visualization and final edition was performed in FigTree v1.3.1. Tajima's Relative Rate Tests [25] were conducted in MEGA6 [26].
3 Results
3.1 Identification and expression of a new MT isoform in African clawed frog tadpoles and adult tissues
A research for MT genes on the S. tropicalis genome assembly release v4.1 at the Joint Genome Institute Genome Portal (JGI Genome Portal) enabled the retrieval of two gene sequences, one showing 100% identity with the X. laevis MT gene sequence identified by Saint-Jacques and Séguin [18] and called MTA (gi|462659|sp|Q05890|MT_XENLA), and another not previously identified as mRNA sequence, located on the scaffold_270:89638-91155, transcript ID 356999. This sequence exhibits all the features to be considered an active coding region. A further search in the database of Expressed Sequence Tags (dbEST) at the NCBI portal led us to the identification of a cDNA fragment (da89f03.y1) from the X. laevis tadpole stage 24 EST (LIBEST_004113) library, whose sequence corresponds to the novel MT isoform identified on the clawed frog genome. In Fig. 1A and B are reported the alignments between the nucleotidic and amino acidic sequences of the two different X. laevis MTs, respectively. The two nucleotidic sequences share about 71% identity. The alignment shows some gaps and the lengths of the coding sequences are different: 186 nucleotides, corresponding to 62 amino acids, for the first identified MT, namely MTA [18], and 180 nucleotides for this new isoform, corresponding to 60 amino acids. However, the most significant differences are highlighted by the amino acid alignment (Fig. 1B). The amino acidic sequence of this new MT isoform shares only 58.1% identity and 64.5% similarity with the X. laevis MTA, and shows unexpected diversity in the sequence, such as the presence of 17 cysteine residues instead of the canonical 20 cysteines of vertebrate MTs, as well as the clustering of some of these cysteine residues into Cys–Cys–Cys sequences instead of the Cys–X–Cys, Cys–Cys and Cys–X–Y–Cys motifs typical of the 20 cysteine residues of vertebrate MTs. For this reason, we termed this atypical MT isoform XlMT-C17.
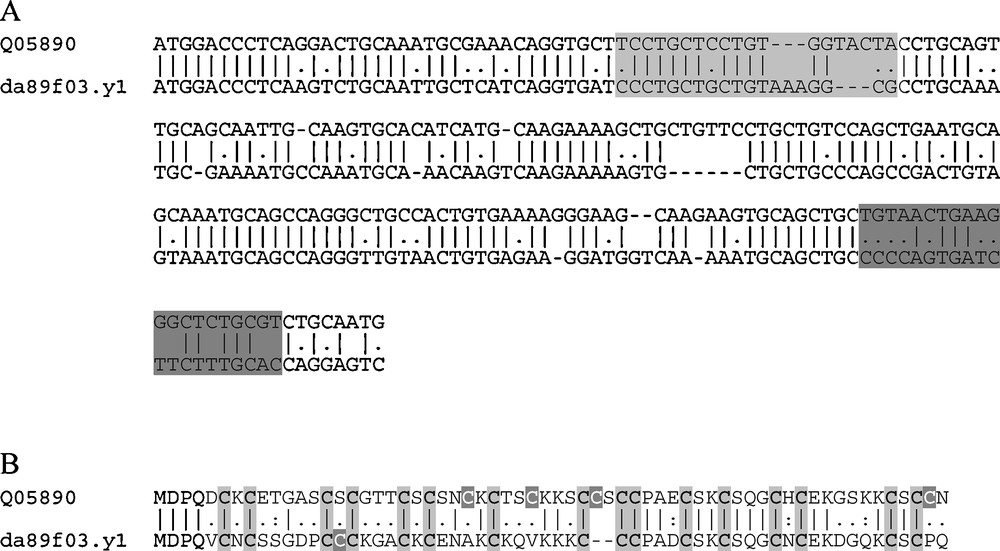
Nucleotidic and aminoacidic alignment of the 2 X. laevis MT isoforms. In the nucleotidic alignment (A), the MT termed Q05890 corresponds to the MTA mRNA sequence identified by Saint-Jacques and Séguin [18], the MT termed da89f03.y1 corresponds to the cDNA fragment from the X. laevis tadpole stage 24 EST (LIBEST_004113) library. The grey boxes indicate the most divergent regions between the two sequences, used to design the primers for conventional and Real Time PCR analyses; specifically, light grey box indicates the region used to design the forward primer, the dark grey box the region used to design the reverse primer. In the aminoacidic alignment (B), light and dark grey highlighted the conserved and unconserved cysteines, respectively. Masquer
Nucleotidic and aminoacidic alignment of the 2 X. laevis MT isoforms. In the nucleotidic alignment (A), the MT termed Q05890 corresponds to the MTA mRNA sequence identified by Saint-Jacques and Séguin [18], the MT termed da89f03.y1 corresponds to ... Lire la suite
As no previous data are available on the effective expression of this new MT form in the African clawed frog tissues, we performed a Real-Time PCR analysis on transcripts from both whole tadpoles (at stage 24 and 48) and adult skin and liver tissues. Specific primers for the XlMT-C17 isoform were designed on the most divergent sequence tracts than MTA isoform (Fig. 1A). To test the efficiency and specificity of the two designed primers, a conventional RT-PCR analysis was performed starting from the total RNA extracted from the stage 24 tadpoles. The PCR reaction produced a single cDNA fragment of the expected length of 159 bp (Fig. 2). The cDNA sequence was 100% identical to that found in the X. laevis tadpole stage 24 EST library.
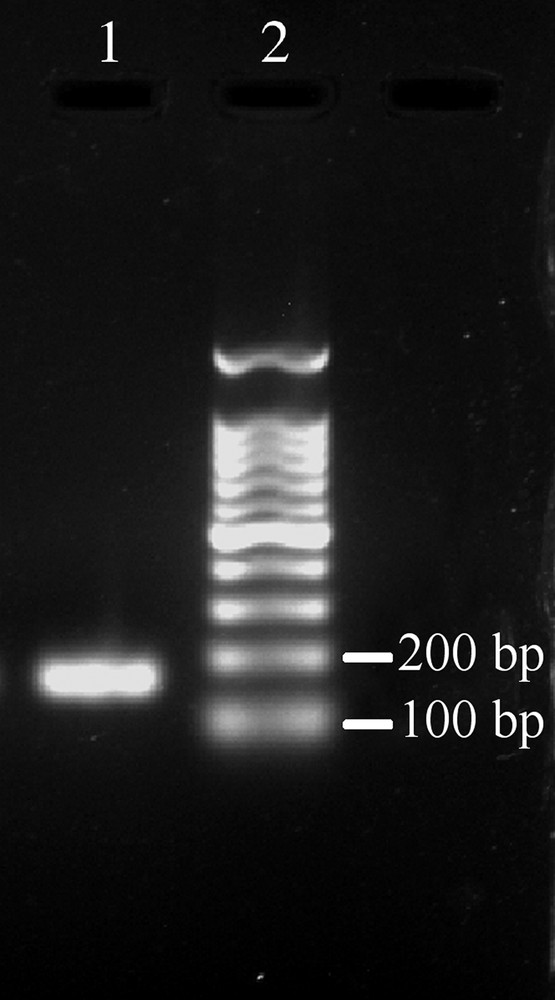
Detection of the atypical MT (da89f03.y1) in X. laevis stage 24 tadpoles by conventional RT-PCR. The RT-PCR was carried out as described in the Section 2, and the resulting products analysed on a 1.2% agarose gel stained with ethidium bromide. The 159 bp band corresponds to the atypical MT cDNA fragment, hereafter called XlMT-C17. Lane 1: RT-PCR product from X. laevis stage 24 tadpoles; lane 2: 100 bp DNA ladder.
The Real Time-PCR revealed the presence of a conspicuous amount of transcripts in both tadpoles and tissues. In particular, in the tadpoles, the levels of this new MT mRNA were lower than adult tissues and remained unchanged between stages 24 and 48 (Fig. 3A), whereas in the two tissues examined high transcript levels were determined, especially in the liver where a huge amount of XlMT-C17 mRNA was found (Fig. 3B). In detail, the amount of XlMT-C17 mRNA transcripts in liver was about three-hundred-fold higher than tadpoles and about three-fold higher than dermis.
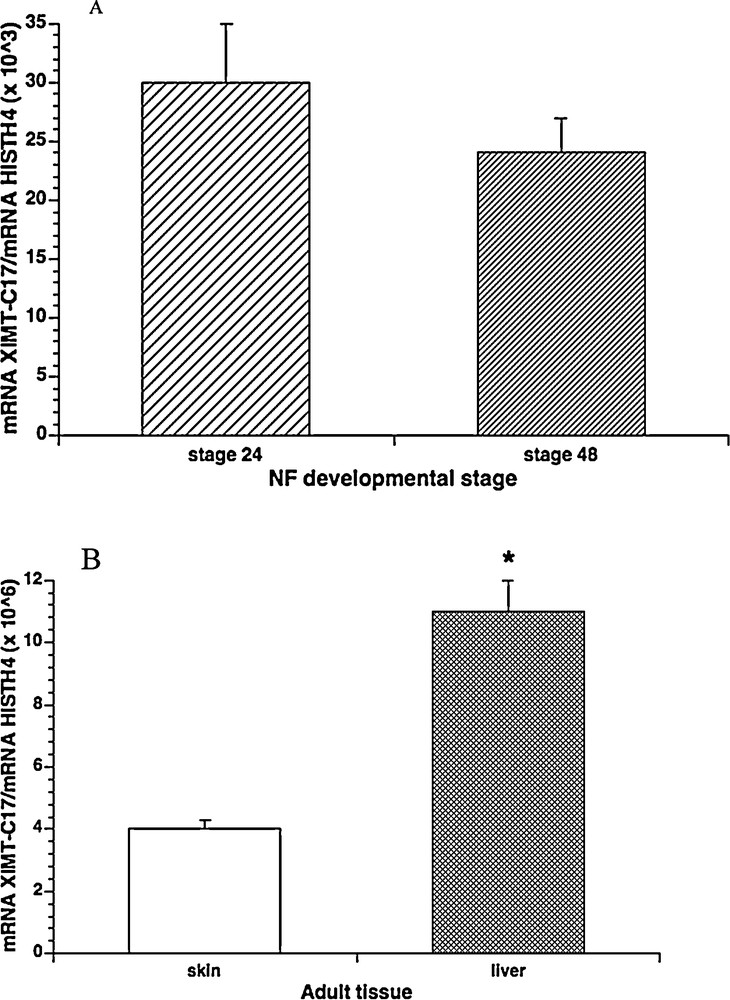
Real Time-PCR for XlMT-C17/Histone H4 ratio in X. laevis developmental stages and adult tissues. A. MT mRNA concentration did not showed statistically significant differences between developmental stages 24 and 48 (P > 0.05). B. MT mRNA concentration showed statistically significant differences between skin and liver tissues (P < 0.0001). Significant differences were found also between each adult tissues and tadpoles (P < 0.0001). The data represent the mean ± s.e.m. (in A, three biological replicates each consisting of three pooled tadpoles; in B, n = 3). Masquer
Real Time-PCR for XlMT-C17/Histone H4 ratio in X. laevis developmental stages and adult tissues. A. MT mRNA concentration did not showed statistically significant differences between developmental stages 24 and 48 (P > 0.05). B. MT mRNA concentration showed statistically significant ... Lire la suite
3.2 Maximum likelihood tree of vertebrate metallothioneins
In order to better investigate the relationships of this new clawed frog MT sequence with the better-known MT sequences from vertebrates, we performed a phylogenetic analysis. Forty-four amino acid sequences representative of almost all vertebrates groups were aligned and used to infer a phylogenetic tree that characterized the evolutionary lineages among the MT genes. The dataset included the atypical human MT1b, characterized by 21 cysteine residues [10]. The maximum likelihood tree (Fig. 4) showed two major clades, one of MTs from teleosts and another comprising the MTs of tetrapods together with the two MT sequences so far identified in cartilaginous fish. In turn, the latter is divided into two clades, one of which, made of the MTs from sharks and the avian MT1 and mammalian MT4 isoforms, emerges basal to the other groups of tetrapod MTs. The new MT-C17 isoform identified in X. laevis merges in the anuran clade, but is the most derived sequence among tetrapod MTs. In addition, Tajima's Relative Rate Test suggests a different evolutionary rate between the two X. laevis MTs (P = 0.021). The human MT1b segregates in the mammalian MT1/MT2 clade (Fig. 4).
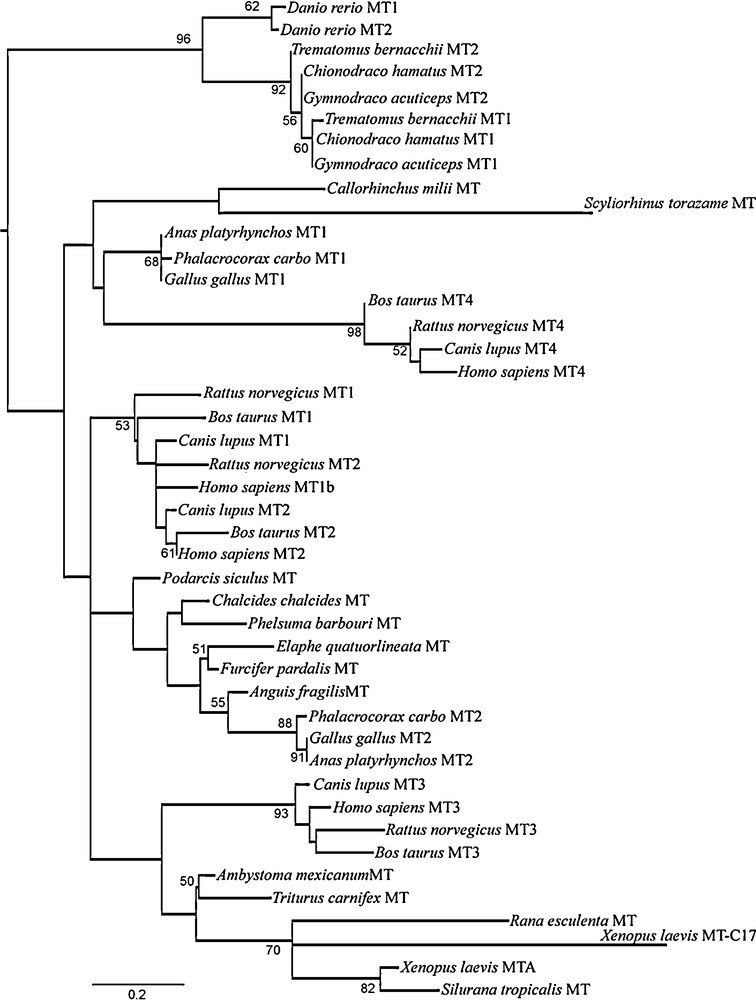
Phylogenetic tree of the amino acid sequences of the vertebrate MTs. The tree was inferred by the Maximum Likelihood (ML) method using the Dayhoff + I + G substitution model implemented in the software SeaView v. 4.0. At the nodes are given values of ML non-parametric bootstrap (cut off > 50%).
4 Discussion
During the last past years, the genome sequencing of model organisms represented an important step towards understanding genes structures and functions; the sequencing is also a valuable shortcut, helping scientists find genes much more easily and quickly. However, genes identified in the genome could not be expressed at all, or may have a differential expression in time and space.
The presence of a gene encoding an atypical MT isoform in the Xenopus genome does not ensure its effective expression. The identification of a corresponding cDNA fragment in the X. laevis tadpole stage 24 EST suggested its expression, which could be limited to the larval phase of the clawed frog life cycle. Our results demonstrate that this atypical MT, to which we gave the acronym XlMT-C17, showing fewer cysteines than any other tetrapods MT isoforms (17 vs. 20, respectively) and two anomalous cysteine triplets, is constitutively expressed throughout the life cycle of Xenopus; moreover, this gene is highly expressed in the liver, the major site of MT expression and synthesis in vertebrates.
In Metazoans, MTs with one or more Cys–Cys–Cys triplets have been found in oligochaetes, mollusks and crustaceans [27–31]. Showing ability for preferentially binding Cu ions [32], these atypical forms of MT in mollusks and crustaceans have been associated with the presence of hemocyanin for oxygen transport, suggesting that these atypical MTs are involved in copper homeostasis associated with the synthesis and degradation of hemocyanin. Indeed, an MT isoform characterized by the presence of three cysteine triplets was isolated from Crassostrea virginica hemocytes [29]. Noteworthy, we determined in the liver of the anuran Rana esculenta a high copper content, about tenfold higher than the amount found in the liver of the lizard Podarcis muralis inhabiting the same geographical area [33]. On the other hand, evidence exists that metal specificity of MT isoforms was achieved by non-chelating amino acid residues in the protein backbone [34]. Thus, at the moment, we cannot state that a correlation exists between the presence of Cu ions and this atypical MT isoform.
Recently, spectroscopic and spectrometric studies performed on the recombinant MT with two uncommon Cys–Cys–Cys triplets identified in the blue crab Callinectes sapidus demonstrated that all cysteine residues participate in the binding of Cu, Zn, and Cd ions, indicating that both Cys triplets act as ligands. NMR analysis reaffirmed the presence of a two-domain structure, each domain containing one Cys triplet and encompassing either the three-metal or the four-metal thiolate cluster [31]. Circular dichroism and UV spectra performed on the β-domain of the human metallothionein 1b demonstrated that the additional cysteine positioned to form the cysteine triplet characterizing this isoform did not perturb the overall structure of the protein or the formation of the metal–thiolate clusters [35]. Despite the presence of two cysteine triplets, the atypical Xenopus MT shows a significantly lower number of cysteines (17 vs. the 20 and 21 cysteines of crab and human MTs, respectively); hence, the XlMT-C17 would not possess the same features. For this very reason, further investigation will be required to identify appropriate gene inducers, assess best ligands and define the functional and structural characteristics of this uncommon amphibian MT.
Finally, as regarding the phylogenetic analysis, our results demonstrate that the MTs from the cartilaginous fish are well separated from the other piscine MTs, clustering together with tetrapods. The MTs from the elephant shark Callorhinchus milii and from the tiger shark Scyliorhinus torazame are quite different from each other: the first is made of 62 amino acids [36], the second of 68 amino acids [37]. In both sequences, the positions of the Cys residues are well conserved and align to other fish and mammalian sequences; noteworthy, the third Cys from the C-terminus shows the typical position found in tetrapod MTs, whereas in MTs from bony fish this cysteine residue is missing.
The phylogeny of the MT isoforms in tetrapods suggests that among them, the mammalian MT4 and avian MT1 isoforms are the most ancestral, as previously reported [9,13], and that the mammalian MT3 clusters forming a sister group of the amphibian MT clade. Our phylogenetic results also demonstrates, as recently suggested [11], that the current nomenclature for distinguishing among different mammalian MT types is not very accurate, being the two MT1 and MT2 not in respectively monophyletic clades, and that differences in gene expression and protein function among mammalian MT isoforms are accompanied by a marked phylogenetic distinction.
At present, unfortunately the low phylogenetic signal of the MT coding regions [38] and [11] does not allow us to best resolve the phylogeny of this gene(s) within the highly heterogeneous group of vertebrates.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.