

1 Introduction
Plant-derived secondary metabolites are an important source of pharmaceuticals and have been expanded beyond its means [1]. Plant-derived secondary metabolites have been used for various other purposes, such as perfumes, pigments, aromatics, pesticides, growth regulators, vitamins, spices, and enzymes. Many of the plant-derived materials are used for drug development, and investigations are being carried out to identify the relationship between chemical structure and activity. Research for the mass production of such compounds is progressing every day [2,3]. In general, secondary metabolites are produced in many parts of the plant in response to external stress. About 300,000 kinds of medicinal plant-derived secondary metabolites are generated worldwide every year and are being extracted and isolated from 1500 plant species. Among those plant species, only 300 species are estimated to produce useful materials based on their physiological activity [4].
Conventional methods of isolation of useful plant compounds are being limited by several factors. The quality of useful plant compounds may tend to vary according to the environment and harvest conditions [5]. Slow growth rate and synthesis of physiologically active substance produced in very small amounts [6,7] and confined to only in certain organs like root, leaves, and flowers further limit the isolation of useful substances from plant systems [8,9]. Therefore, it is highly desirable to develop appropriate new product development processes. These processes can be achieved by the development of in vitro mass production systems, such as plant tissue and cell culture techniques. The mass production of secondary metabolites is reliable and almost not subject to the influence of the environment, and production of useful products can be possible in a short period of time [10,11].
Catharanthus contains highly functional and bioactive alkaloids, from which over 130 have been isolated and identified [12]. In particular, pharmacologically important secondary metabolites, such as vinblastine, vindoline, vincristine, ajmalicine, and catharanthine are being used in the treatment of acute leukemia, malignant lymphoma, and Hodgkin's diseases. Extraction of bioactive compounds directly from plant parts pose an economic burden due to very slow growth, and contents of bioactive compounds are present in very small amounts and only limited to specific organs. Because of their complex chemical structure, their chemical synthesis is difficult to obtain. Important secondary metabolites of Catharanthus, vinblastine and vincristine are present in very small amounts in the plant (approximately 0.0002% of fresh weight) [13]. In order to obtain 1 g of vincristine, more than 500 kg of Catharanthus are required, which is commercially unviable and constitutes an economic burden. To overcome these setbacks, various plant cell culture systems are being used in alkaloid production and tissue culture studies, such as hairy root culture, suspension culture, and gene transfection studies have been conducted [12,14,15]. The first successful Catharanthus tissue culture system was reported by Barun [16]; the callus cultures were reported by Badcock and Carew in 1962 [17]; and the suspension culture was studied by Harris et al. [18]. However, such plant cell culture techniques, as a system of economically important secondary metabolites production in Catharanthus, have not reached the level of mass production yet, and there is a need for the development of systematic tissue culture methods for the mass production of economically important secondary metabolites from Catharanthus.
One of the disadvantages of the use of callus or DDCs-derived culture is that cells may lose their mitotic activity after a prolonged culture period [19,20], and the production of commercially important materials is highly variable due to genetic instability. In order to overcome these problems, much recently, the use of undifferentiated tissues from plant meristem was reported [21]. Meristematic tissues are the source of undifferentiated stem cells and produce various kinds of proliferating cells, which eventually generates the various kinds of plant tissues [22]. Recently, a significant increase in the knowledge about the cellular and molecular mechanisms involving functional meristems has been seen [23,24]. Plant stem cells, which are embedded in meristematic tissues, located at the tips of roots, shoots, and/or in the vascular system, can multiply and give rise to a variety of cells that eventually undergo a differentiation process and simultaneously produce new stem cells [25]. The use of these meristematic tissues in tissue culture techniques avoids the dedifferentiation process. Furthermore, these meristem cells have been reported to have the self-organization, special mechanism for preventing mutations in addition to the multipotential proliferative and indeterminate growth capacity [19,20]. Taking advantage of meristematic cell characteristics, such as innately undifferentiated growth pattern, stable growth rate, and retention of all the characteristics of the parent plant without mutation, the stable mass production of useful materials can be easily achieved [21].
In the present investigation, we aimed at isolating and characterizing CMCs from Catharanthus stem and establish stable cell culture from CMCs rather than from conventional callus tissue cultures, which are derived as a result of dedifferentiation. We identified differential growth characteristics of CMCs and DDCs and standardized the suitable in vitro conditions for their prolonged culture period, and also partially identified the suitable conditions for increased synthesis of TIAs.
2 Materials and methods
2.1 Plant material
Catharanthus seeds were surface sterilized with 70% ethyl alcohol for 1 min, followed by 1% sodium hypochlorite for 15 min, and rinsed 5 to 10 times with sterilized distilled water, and then seeds were air dried on clean filter paper. Seeds were cultured in Murashige Skoog (MS) medium with 0.65% agar agar; pH was adjusted to 5.8 before autoclaving. After inoculation, the seeds cultures were kept under dark conditions for around 3–5 days for induction of germination. After seed germination, cultures were maintained under the 16-h light period and at a temperature of 25 °C.
2.2 Isolation of meristematic stem cells
Meristematic cells were obtained from 30-day-old in vitro seedlings of Catharanthus stems. We gently dissected the Catharanthus stem segments obtained from the in vitro seedlings using sterilized pincette and blades (No. 11). The isolated tissue samples contained epidermis, endodermis, phloem, xylem, cambium, cortex and pith tissues. Xylem and pith tissue were gently removed, and the absence of xylem and pith tissues was confirmed by staining the tissues with phloroglucinol–HCl, where lignin deposition was detected by the development of a red color (Fig. 1A).

Comparison of morphologies of cambial meristematic cells (CMCs) and dedifferentiation-derived callus (DDCs) at different stages of induction and regeneration. A. Cross and longitudinal sections of Catharanthus stem stained with xylem and pith specific dye, phlorogluciol–HCl observed under dark filed microscope (Eclipse 80i, Nikon, Japan). B. Isolated cambium meristem cells cultured on induction media. C. Growth of CMCs after 7 days of induction. D. Growth of CMCs after 10 days of induction. E. Initiation of DDCs after 15 days of induction (dark yellow color margins indicated with arrow marks). F. Growth of CMCs (top arrow mark) and DDCs (bottom arrow mark) after 30 days induction, where CMCs and DDCs were clearly distinguishable. The tissues were observed under a stereomicroscope (SZX12, Olympus, Japan). For interpretation of the references to color in this figure caption, the reader is referred to the online version of this article.
2.3 Differential induction of CMCs and DDCs
The isolated tissues were transferred onto the differential induction medium containing Gamborg's B5 salts and vitamins (Duchefa, The Netherlands) (3.16 g·L−1), sucrose (30 g·L−1), agar agar (6.5 g·L−1), and different combinations of NAA and kinetin (Fig. 1B, Table 1); the pH of the medium was adjusted to 5.7. The medium was autoclaved at 121 °C for 15 min. The cultures were maintained in dark conditions, and in regular intervals cultures were observed for differential inductions of CMCs and DDCs on different induction media. After differential induction, CMCs and DDCs were separated and grown on suitable media. Cultures were maintained at 25 °C in dark conditions and subcultured for every 2 weeks.
Combination of NAA and kinetin used (T1–T16) in the Catharanthus cell cultures.
| NAA (mg·L−1) | Kinetin (mg·L−1) | |||
| 0.5 | 1 | 1.5 | 2 | |
| 0.5 | T1 | T2 | T3 | T4 |
| 1 | T5 | T6 | T7 | T8 |
| 1.5 | T9 | T10 | T11 | T12 |
| 2 | T13 | T14 | T15 | T16 |
2.4 Histological observation
Histological observations were made using both light and confocal microscopy. For histological studies with light microscopy, the paraffin section was used. The steps of paraffin block technique included fixation, dehydration, and paraffin embedding; 30-day-old tissues from CMCs and DDCs were fixed with a solution containing formalin acetic acid-alcohol (FAA), 10% of formalin, 5% of glacial acetic acid, and 50% of ethyl alcohol. The samples were incubated for 16 h at 4 °C, and then washed with 95% ethyl alcohol. For dehydration, tissues were treated with series of solutions containing different concentrations of ethyl alcohol and of tertiary butyl alcohol (TBA). The samples were incubated for 60 min in a solution containing 50% ethyl alcohol (95%) and 10% TBA, 60 min in solution containing 50% ethyl alcohol (95%) and 20% TBA, 60 min in solution containing 50% ethyl alcohol (95%) and 35% TBA, twice for 60 min in a solution containing 50% ethyl alcohol (100%) and 50% TBA, and 60 min in a solution containing 25% ethyl alcohol (100%) and 75% TBA. Finally, samples were completely dehydrated by incubating in 100% TBA for 60 min twice. For tissue embedding, the tissue samples in the TBA were shifted to an oven (60 °C), and then were added paraplast (Leica instruments, Germany) in a quantity equal to that of TBA. After melting the paraplast, TBA was allowed to volatilize. After volatilization of TBA, tissues were treated with fresh paraffin solvent twice for 1 h in 60 °C. Tissues embedded in paraffin wax were then sectioned with the rotary microtome (Leica instruments, Germany) and cut in 5–10-μm thin sections. Tissue ribbons were mounted on glass slides by keeping them on a warmer slide (at 45 °C). The glass slides were dried and stored in a slide box.
For general morphological observations by light microscopy, the sections were stained with phloroglucinol–HCl and 0.1% toluidine blue for the comparison of CMCs and DDCs. The staining technique included deparaffinization, rehydration, staining and dehydration, and clearing. The slides were deparaffinized in xylene for 10 min, and this was repeated three times. The samples were rehydrated in ethanol series (100% to 30%), and stained in 0.1% toluidine blue, and phloroglucinol with 20% HCl for 1 h, and then washed in water to remove excess dye. The slides were then dehydrated in ethanol and mounted with Canada balsam for examination.
We used the fresh CMCs and DDCs for the fluorescence microscopy study; the fluorescence images of plant samples were captured with a confocal laser scanning microscope (Olympus Fluoview FV1000 version 2.0) and FV10-ASW 2.0 viewer software. The fluorescence emissions were collected using an Ar-ion laser, green fluorescence signal at 488 nm (30 mW) and a He–Ne laser, yellow fluorescence signal at 543 nm (1 mW), and a red fluorescence signal at 633 nm (10 mW) illumination. The pinhole was used in the imaging system with a z-scan mode to make images from different layers, and stacks of images were collected every 2 μm along the z-axis.
2.5 Expression analysis of TIAs biosynthesis and developmentally regulated genes
2.5.1 Total RNA isolation and cDNA synthesis
Catharanthus meristematic cells were collected from 10-days-, 30-days-, and 50-day-old in vitro grown cultures. Four tissue samples were collected from each one of the CMCs and DDCs, which were maintained on their respective media. Total RNA was isolated using the RBC plant RNA isolation kit following the manufacturer's instructions. The RNA concentration and purity were measured with a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies, USA). The RNA integrity was checked by formaldehyde gel electrophoresis. Six hundred nanograms of total RNA was used for reverse-transcription with a Rocket script RT premix kit (Bioneer, South Korea). First-strand cDNA synthesis was carried out in a final volume of a 20-μL reaction mixture using oligo-dT (20 mer) primer according to the manufacturer's protocol. cDNA synthesis was carried out at 30 °C for 1 min, at 60 °C for 1 h, and at 95 °C for 5 min. The cDNA was diluted twice with nuclease-free water and stored at −20 °C until the quantitative reverse-transcription PCR (qRT-PCR) experiment.
2.5.2 qRT-PCR analysis of CMCs and DDCs
For the determination of expression patterns of Phloem intercalated with xylem (PXY) and Wuscehl related Homeobox 4 (WOX4) genes, which are known to be involved in the proliferation of procambial/cambial stem cells, they were identified by BLAST searches with Arabidopsis PXY (NM_125541) and WOX4 (AY251396) genes against the Medicinal Plant Genomics Resource (http://medicinalplantgenomics.msu.edu/). To know the activity of the TIAs biosynthetic pathway genes, such as tryptophan decarboxylase (TDC), geraniol 10-hydroxylase (G10H), stritosidine synthase (STR), desacetoxyvindole 4-hydroxylase (D4H), and acetyl-CoA:4-O-deacetylvindoline (DAT) [26,27], qRT-PCR analysis was carried out in cultured Catharanthus cells.
qRT-PCR primers for PXY, WOX4, TDC, G10H, STR, D4H, and DAT genes were designed using Primer 3 software program (http://frodo.wi.mit.edu/primer3/) with default parameters. Primers used in the present study are listed in Table 2. Elongation factor-1-alpha (EF1a) was used as an internal control. In order to check the primers specificity, regular PCR was performed, and the products were analyzed by electrophoresis on 1.5% agarose gels. Further, the specificity of primers was verified by a melting curve analysis. The qRT-PCR reaction was performed under the following conditions; 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 62 °C in Bio-Rad CFX 96 real-time system (Bio-Rad, USA). The final volume for all reactions of qRT-PCR was 25 μL, and each reaction mixture included 12.5 μL of 2X green star qPCR master mix (Bioneer, South Korea), 2 μL of primer mixture (10 pM each), 2 μL of diluted cDNA, and 8.5 μL of DEPC water.
qRT-PCR Primers used in the present investigation.
| Primer name | Accession | Data base | Primer sequence | Amplicon size (bp) |
| EF1a-F | EU007436 | NCBI | TCAGGAGGCTCTTCCTGGTGA | 115 |
| EF1a-R | AGCTCCCTTGGCAGGGTCAT | |||
| PXY-F | cra_locus_6307_iso_1_len_3407_ver_3 | MPGR | TGATAAGCCAGGAGGGAAT | 161 |
| PXY-R | TCCCGCCTGATTTCAGATAC | |||
| WOX4-F | cra_locus_8377_iso_1_len_1147_ver_3 | MPGR | GGACTTTATGATTTAGTACACGTCTC | 122 |
| WOX4-R | CCCTACAAGATCAATACATATGCAAC | |||
| TDC-F | M25151 | NCBI | TCCGAAAACAAGCCCATCGT | 126 |
| TDC-R | AAGGAGCGGTTTCGGGGATA | |||
| G10H-F | AJ251269.1 | NCBI | TAGCAGGGACGGACACAACATCAA | 304 |
| G10H-R | TCACGTCCAATTGCCCAAGCATTC | |||
| STR-F | X61932 | NCBI | TGACAGTCCCGAAGGTGTGG | 122 |
| STR-R | CGCCGGGAACATGTAGCTCT | |||
| D4H-F | U71605.1 | NCBI | TACCCTGCATGCCCTCAACC | 121 |
| D4H-R | TTGAAGGCCGCCAATTTGAT | |||
| DAT-F | AF053307 | NCBI | CTTCTTCTCATCACGTACCAACTC | 172 |
| DAT-R | ATACCAAACTCAACGGCCTTAG |
2.6 Analysis of TIAs by high-performance liquid chromatography (HPLC)
The extraction procedure was adopted from Tikhomiroff and Jolicoeur [28]. Briefly, CMCs and DDCs samples from 50-day cultures were lyophilized overnight and grounded in a mortar, followed by extraction with methanol for 1 h in a sonicating bath. The extracts were centrifuged at 15,000 g for 5 min at ambient temperature, and the supernatant was filtered through a 0.45-μm filter (Whatman, USA) and collected into a glass HPLC vial for analysis. Stock solutions of vindoline, vinblastine, vincristine, and catharanthine were prepared at concentration of 5 mg·mL−1 in DMSO, and stored at −20 °C. Individual stock solutions were prepared in different amounts for the preparation of calibration graphs, linear in the range of the working concentration (0.03 ∼ 1.25 mg·mL−1). HPLC analysis, taken from Tikhomiroff and Jolicoeur method [28] with certain modifications, performed using a Agilent 1100 series (Agilent Technologies, USA), binary pump model DE43618438 (Agilent Technologies, USA), UV detector DAD system model G1315B (Agilent Technologies, USA). An Eclipse XDB-C18 column—4.6 mm ID × 250 mm (5 μm) (Agilent Technologies, USA)—coupled with a security guard column (Phenomenex, USA) was used at a column temperature of 35 °C with UV detection at 256 nm and a constant flow rate of 1 mL/min for all the analysis process. The mobile phase consisted of 5 mM Na2HPO4 (pH adjusted to 6 with H3PO4) (solvent A) and acetonitrile (solvent B), from which solvent A was filtered with a glass vacuum filter with a PVDF membrane filter (Merk Milipore, USA). The volume ratio (A:B) was 0–20 min, linear gradient from 65:35 to 20:80; 20–25 min, linear gradient from 20:80 to 0:100; 25–35 min, isocratic elution with 0:100 (v/v) and 35–40 min, isocratic elution with 65:35 (v/v) for equilibration.
3 Results
3.1 Establishment of cambial meristematic cell lines
For the establishment of the CMCs and DDCs, Catharanthus cambial meristematic tissues were grown on media containing different combinations of hormones (Table 1). Cell proliferation was observed with white and transparent tissue from the surface of the explants after 4 to 10 days of inoculation (Fig. 1C and D). The characteristics of the transparent and white tissue induced from the surface of the cell layer differed slightly depending on the composition of the hormones (with the exceptions of T12, T14, T15, and T16). CMCs were induced within 4 to 7 days of culturing in media with equal concentrations of the NAA and kinetin combinations (T1, T6, and T11), as well as in media with lower concentration of NAA than of kinetin (T2, T3, T4, T7, and T8). After 2 to 3 weeks of inoculation, the surface of the explants was completely covered with white and transparent tissues (CMCs), and from the margins of the white and transparent tissue, light yellow-colored DDCs were induced (Fig. 1E). After 4 weeks, CMCs and DDCs could be clearly distinguishable: white transparent cell layers derived from CMCs and brownish-yellow-colored cells derived from DDCs (Fig. 1F). After 30 days of culturing, CMCs could be gently separated from the DDCs, which are supposed to be derived from pericycle, phloem, cortex, and epidermis tissues [29].
The influence of hormones on the growth of two cell lines is determined by their differential growth in media containing different combinations of NAA and kinetin. Following the separation of two kinds of cells, CMCs and DDCs were transferred onto T1, T3, T9, and T11 media. The white and transparent cell characteristic of CMCs was maintained even after several rounds of subculturing (8–10 subcultures) (Fig. 2A), whereas the DDCs’ color changed from yellow to brown after two subcultures (Fig. 2B). Interestingly, we also observed root induction from DDCs (Fig. 2C). The initial growth of the cells in media containing low concentration of NAA than kinetin or equal concentration of NAA and kinetin is relatively higher and fast. However, the growth rate of CMCs is high in media containing higher concentrations of NAA than of kinetin, and the growth rate of DDCs is higher on media containing lower concentrations of NAA than of kinetin.
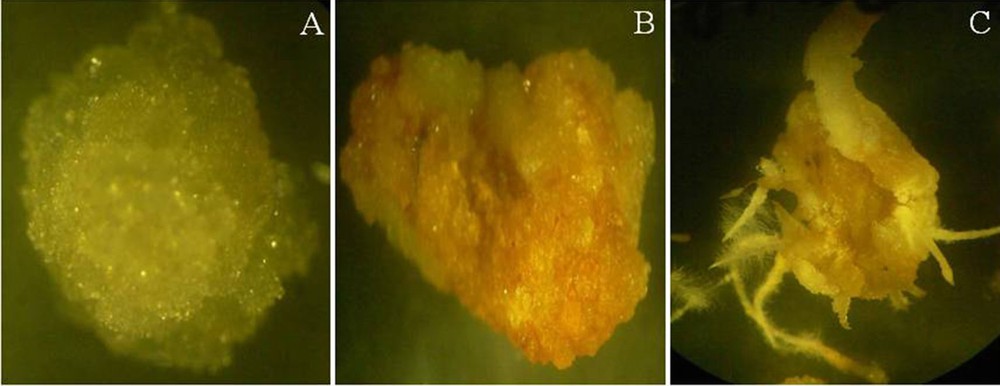
Isolated cambial meristematic cells (CMCs) and dedifferentiation-derived callus (DDCs) grown on specific induction media. A. CMCs are white/transparent, with a globular shape. B. DDCs are brown in color, with a friable nature. C. Induction of root from DDCs. The tissues were observed under a stereomicroscope (SZX12, Olympus, Japan). For interpretation of the references to color in this figure caption, the reader is referred to the online version of this article.
3.2 Histological observation of CMCs and DDCs
Xylem and pith tissues were separated from cambial tissue based on their dark-red color in staining with lignin-specific dye phloroglucinol–HCl (Fig. 1A). Cell lines derived from the CMCs and DDCs were clearly distinct (Fig. 3A and B). The cell line derived from the cambium, the CMCs showed no reaction to the dye phloroglucinol–HCl (Fig. 3C), whereas DDC specific cell line micro-sections stained red-violet in color (Fig. 3D). Thus, this result suggests that DDCs with lignin deposition in cell walls were derived from the phloem, cortex, and epidermis tissues. In addition, the primary cell walls were stained with purple, and the secondary cell walls were stained blue with toluidine blue. Under the microscope study, the CMCs micro-sections were characterized by a purple color (Fig. 3E), whereas DDCs micro-sections were characterized by a blue color (Fig. 3F). This result indicates the CMCs with primary cell walls, and the DDCs with the secondary cell wall. Thus, the main difference in morphology of the cell wall was confirmed. The physical features of the CMCs are primary thin cell walls and small and compact cell size. DDCs with secondary cell walls are thick, and the size of the cell was slightly larger in comparison with that of the CMCs. CMCs and DDCs could also be distinguished based on their distinct autofluorescence characteristics under the confocal laser microscope. Excitation CMCs and DDCs at 543 and 633 nm resulted in strong autofluorescence signal only in DDCs, whereas low fluorescence was observed in CMCs. However, fluorescence was much stronger at 543 than at 633 nm (Fig. 4A–H).
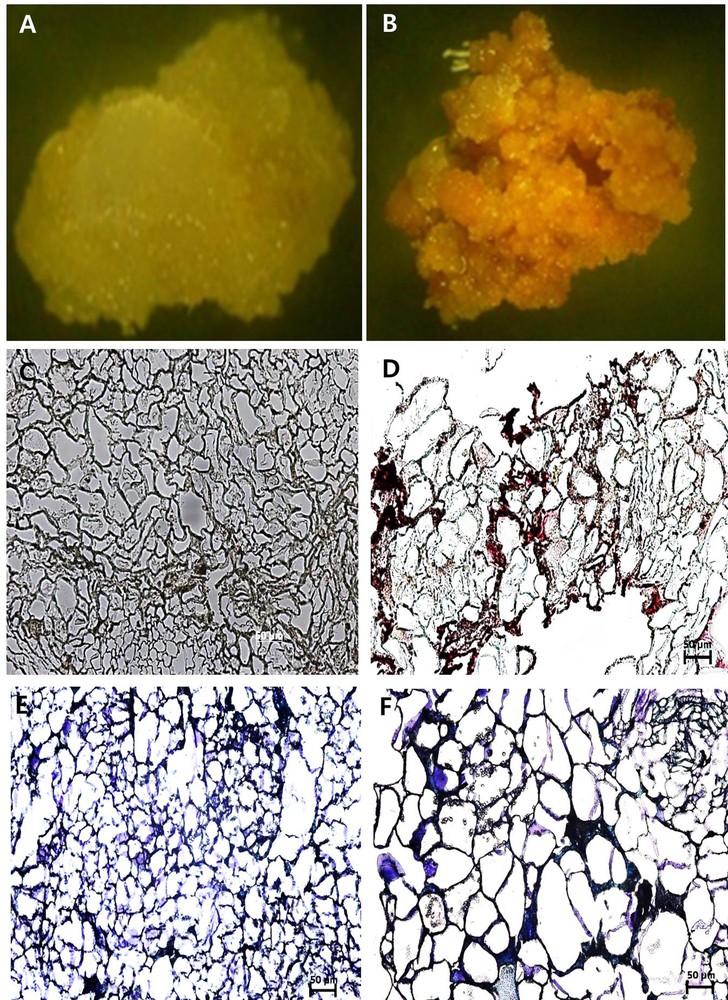
(Color online.) Optical micrographs of cambial meristematic cells (CMCs) and dedifferentiation-derived callus (DDCs) with paraffin micro-sections stained with phloroglucinol–HCl reagent and toluidine blue. A and B. CMCs and DDCs observed under a stereomicroscope, respectively. C and D. CMCs and DDCs stained with phloroglucinol–HCl as a reagent, observed under a dark-field microscope (Eclipse 80i, Nikon, Japan), respectively, at 20×. E and F. CMCs and DDCs stained with toluidine blue observed under a dark-field microscope (Eclipse 80i, Nikon, Japan), respectively, at 20×. The scale bar equals 50 μm.
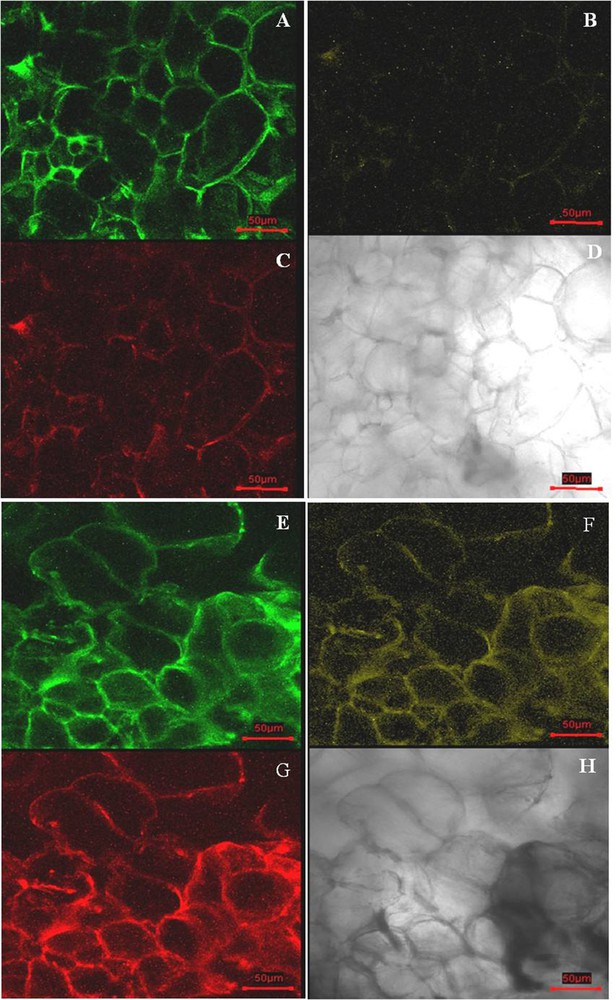
(Color online.) Confocal laser scanning microscopy of fresh cambial meristematic cells (CMCs) and dedifferentiation-derived callus (DDCs) samples. CMCs exposed to 488-nm (A), 543-nm (B), and 633-nm (C) lasers, respectively; optical micrograph of CMCs (D); DDCs exposed to 488-nm (E), 543-nm (F), and 633-nm (G) lasers, respectively; optical micrograph of DDCs (H). The scale bar equals 50 μm.
3.3 qRT-PCR analysis of DDCs and CMCs
Genetic analyses showed that PXY and WOX4 genes play an important role in promoting the proliferation of procambial/cambial stem cells [30]. In order to check their expression patterns in CMCs and DDCs, we carried out a qRT-PCR analysis. Expression analyses were carried out at three different time intervals; 10 days after induction (CMCs), 30 days after induction (CMCs and DDCs), and 50 days after induction (CMCs and DDCs). After 10 days of culturing, CMCs were induced and showed the maximum expression of WOX4 grown in T9, whereas PXY showed the maximum expression in T1. The least expression of PXY was observed in T9 (Fig. 5A). In 30-day-old CMCs, WOX4 showed similar expression in all treatments, except in T11. In contrast, the expression levels varied slightly in DDCs, although the expression levels were significantly less when compared to CMCs. Maximum expression levels of PYX were observed in CMCs and DDCs grown on T1. The least expression of PXY was observed in CMCs grown in T11 (Fig. 5B). In 50-day-old CMCs, WOX4 showed a maximum expression in T9 with the least expression in T3. DDCs showed the maximum expression of WOX4 in T1 and the least expression in T3 and T9. The expression levels were relatively less in DDCs when compared to CMCs (Fig. 5C). The maximum expression levels of PYX were observed in CMCs and DDCs grown in T1 and T3, respectively. The least expression of PXY was observed in DDCs grown in T11. The expression levels were relatively less in DDCs when compared to CMCs with an exception in the T3 treatment.

(Color online.) Quantitative reverse-transcription-PCR analysis of Wuscehl related Homeobox 4 (WOX4) and Phloem intercalated with xylem (PXY) genes in cambial meristematic cells and dedifferentiation-derived callus grown under different combination of hormones. Expression profiles of WOX4 and PXY genes in (A) 10-day-old samples; (B) 30-day-old samples; and (C) 50-day-old samples. The Elongation factor-1-alpha gene was used as an internal control. Expression fold changes in genes are shown in terms of log2-fold. The error bars indicate ± SD.
To gain a better understanding of the expression levels of TIAs biosynthetic pathway genes, such as TDC, G10H, STR, D4H, and DAT (Fig. 6), gene expression profiles were analyzed in CMCs and DDCs by qRT-PCR. In 10-day-old CMCs, All TIAs biosynthetic pathway genes with the exception of the G10H gene showed maximum expression levels in T1. G10H showed the maximum expression in T11, and least expression in T1. Almost similar expression levels were observed in both cases; TDC and STR, and D4H and DAT genes (Fig. 7A). In 30-day-old CMCs, TDC, STR, D4H, and DAT genes showed the maximum expression in T1 and the least expression in T11. Similar trends were also observed in the case of DDCs, but with lower levels of expression. D4H showed almost similar expression levels in DDCs grown in any media. G10H showed the maximum expression in CMCs when compared with DDCs grown in any media (Fig. 7B). In 50-day-old CMCs, G10H showed the maximum expression in T3 and the least expression in T9. A comparatively higher level of expression of G10H was observed in DDCs grown in any media with exception in T9. TDC showed the maximum expression in T1, and slightly lower levels of expression were observed in other treatments. In DDCs, TDC showed the maximum expression in T3, and similar levels of expression were observed DDCs grown under other treatments. In CMCs, STR showed the maximum expression in T1 and T3 and the lowest expression in T11 (Fig. 7C), whereas significantly lesser expression levels were observed in DDCs. In DDCs, STR showed almost similar expressions levels in cells grown on any media. In CMCs, D4H showed the maximum expression in T9 and the least expression in T11, whereas in DDCs the maximum expression levels of D4H were observed in DDCs grown in T1 and slightly lesser levels in others. In CMCs, DAT showed similar expressions in all treatments. However, in case of DDCs, the maximum expression was observed in T3 and the least expression in T9.

(Color online.) Biosynthetic pathway for terpenoid indole alkaloids in Catharanthus. The genes used in the quantitative reverse-transcription-PCR studies are highlighted.
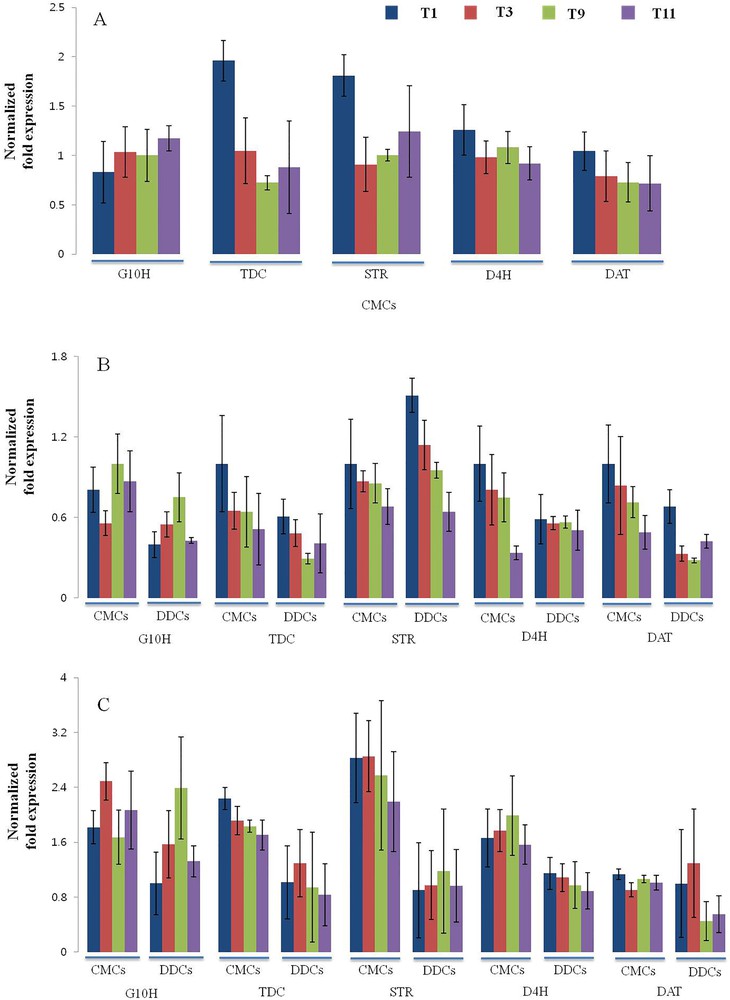
(Color online.) Quantitative reverse-transcription-PCR analysis of terpenoid indole alkaloids (TIAs) biosynthetic pathway genes (TDC, G10H, STR, D4H, and DAT) in cambial meristematic cells and dedifferentiation-derived callus grown under different combination of hormones. Expression profile of TIAs biosynthetic pathway genes in (A) 10-day-old samples; (B) 30-day-old samples; (C) 50-day-old samples. The Elongation factor-1-alpha gene was used as an internal control. Expression fold changes in genes are shown in terms of log2-fold. Error bars indicate ± SD.
3.4 Analysis of alkaloids contents in CMCs and DDCs
The determination of the alkaloid content from CMCs and DDCs was performed by reversed HPLC. The samples were injected to evaluate the separation efficiency of the alkaloids based on a standard mixture of the four alkaloids. Table 3 shows that CMCs improves alkaloid biosynthesis in the culture, especially vindoline, vinblastine, vincristine, and catharanthine. Vindoline was only detectable in cultures grown in T1, T3, T9 of CMCs (0.036 to 0.051 mg·g−1 DW), whereas it was not detected in CMCs grown in T11 and DDCs grown in any of the media. Vinblastine showed a maximum detection in CMCs grown in T9 (0.794 mg·g−1 DW) and the least detection in DDCs grown in T11 (0.479 mg·g−1 DW), whereas in CMCs grown on T11, the content of vincristine reached a maximum (0.279 mg·g−1 DW) and the least detection was in DDCs grown on T3 (0.183 mg·g−1 DW). The catharanthine content was noted to be maximal in CMCs grown on T9, which was about five-fold higher than the least detection in DDCs grown on T11 and minimum in CMCs and DDCs grown on T11.
Effect of NAA and kinetin on accumulation of indole alkaloids in Catharanthus CMCs and DDCs.
| Tissue | Media | Vindoline | Vinblastine | Vincristine | Catharanthine |
| mg·g−1 dry weight | |||||
| CMCs | T1 | 0.051 ± 0.009a | 0.613 ± 0.007c | 0.230 ± 0.004d | 13.77 ± 0.99c |
| T3 | 0.041 ± 0.009a | 0.740 ± 0.025b | 0.268 ± 0.004b | 10.33 ± 0.21d | |
| T9 | 0.036 ± 0.005b | 0.794 ± 0.025a | 0.243 ± 0.004c | 19.40 ± 0.19a | |
| T11 | Ndc | 0.613 ± 0.006c | 0.279 ± 0.006a | 3.98 ± 0.04f | |
| DDCs | T1 | Ndc | 0.582 ± 0.005d | 0.231 ± 0.001d | 14.81 ± 0.37b |
| T3 | Ndc | 0.574 ± 0.024d | 0.183 ± 0.002f | 7.82 ± 0.74d,e | |
| T9 | Ndc | 0.489 ± 0.002e | 0.194 ± 0.002e | 10.65 ± 0.53d | |
| T11 | Ndc | 0.479 ± 0.006e | 0.199 ± 0.003e | 3.88 ± 0.29f |
4 Discussion
Meristematic cells located at the apical regions of shoots and roots or located inside the vascular system are able to divide and generate cells that ultimately undergo specialization or differentiation, while at the same time giving rise to new stem cells [25]. These cells are considered to be immortal due to their ability to divide an unlimited number of times. Cell suspension cultures were believed to proliferate through the dedifferentiation process [31]. However, recent studies suggest that callus formation might not involve a simple backward reprogramming of dedifferentiation process [29]. Regardless of the mechanism involved, callus formation results in the generation of various kinds of specialized cell types [32]. Cell cultures derived from such cellular mixtures often show limited growth with low and inefficient yields of natural products synthesis [11] due to deleterious genetic/epigenetic changes that occur during callus formation [32,33]. In the present study, we isolated an innately undifferentiated cambial cell line from Catharanthus stems, which is distinct from DDCs.
CMCs and DDCs are clearly distinguishable based on morphological features and cell specific gene expression patterns. Growth media with a varied combination of hormones were used to compare the growth properties of DDCs and CMCs isolated from the Catharanthus stem. DDCs showed slow growth and displayed a brownish color in 30 days (after two subcultures), whereas CMCs showed rapid growth even after 3 months of subculture. CMCs with primary cell walls and DDCs tissue with secondary cell walls were confirmed by the observation of marked lignin deposition in cell walls of DDCs-derived from the pericycle, phloem, cortex, and epidermis tissues. The physical features of the CMCs are the thin primary cell wall and small and compact cells size, whereas the physical features of the DDCs are thick secondary cell walls and larger cell size. CMCs and DDCs could also be distinguished based on their distinct autofluorescence characteristics under the confocal laser microscope. DDCs showed strong autofluorescence signals when excited at 543 and 633 nm, whereas significantly low fluorescence was observed in CMCs. Overall, our findings showed that these cell lines displayed stem cell-like properties in agreement with the known CMCs identity [21]. In the present investigation, we observed the development of root-like structures from DDCs possibly derived from pericycle-like tissues [29]. Recently, it has been reported that callus formation from tissues, such as cotyledons, roots, and petals were hindered in mutants, which are not capable of lateral root formation [29]. It suggests that an ectopic activation of lateral root formation is a common mechanism associated with callus formation from multiple organs, and callus is not the result of the dedifferentiation process backward to an undifferentiated state but rather an organization formed from root pericycle-like cells [29]. The PXY gene that encodes for a LRR-RLK protein is reported to be differentially expressed in CMCs cells and DDCs [34,35]. The analysis of qRT-PCR results established that the expression of PXY gene was upregulated significantly in CMCs compared with DDCs. WOX4 gene, which belongs to the WUS subclade in WOX family, is required for promoting the proliferation of cambium stem cells. WOX4 had previously been shown to promote vascular cell division [30] and is required to maintain the cell proliferation activity of vascular stem cells [36]. Consistent with these findings, the expression analysis established that the expression of WOX4 was upregulated significantly in CMCs than in DDCs.
The Catharanthus plant is known to produce over 130 different TIAs compounds, including the powerful cytotoxic drugs vinblastine and vincristine [27]. Catharanthus alkaloids synthesis share many common biosynthetic steps. Critical early steps in the TIAs biosynthesis include the reaction-mediated TDC and STR. The first committed steps of TIAs biosynthesis in the indole and terpenoid precursor branches are catalyzed by TDC and G10H, respectively. STR involves the condensing of tryptamine and secologanin, thereby resulting in the formation of strictosidine, which is a general backbone structure of the TIAs [27,37]. A cytosolic 2-oxoglutarate-dependent dioxygenase enzyme known as D4H is involved in the 4-hydroxylation of desacetoxyvindoline [38]. The final O-acetylation of deacetylvindolineis is catalyzed by the cytosolic DAT enzyme to yield vindoline [39,40]. Almost all stages of CMCs showed the maximum expression of TIAs biosynthesis genes in cells grown on media containing 0.5 mg·mL−1 of NAA and 0.5 mg·mL−1 of kinetin (T1). The HPLC analysis revealed that a relatively higher accumulation of TIA compounds was observed in CMCs than in DDCs. However, the observed higher expression of TIAs pathway biosynthesis genes in CMCs grown on T1 media is not correlated with the higher accumulation of TIA compounds. Cells grown on T9 media containing 1.5 mg·mL−1 of NAA and 0.5 mg·mL−1 of kinetin showed maximum accumulation of TIA compounds. Therefore, T9 would be suitable media for CMCs-derived suspension cultures for increased production of Catharanthus TIAs, whereas, T1 media would be a more suitable for proliferation of CMCs.
In conclusion, we isolated undifferentiated CMCs in Catharanthus and characterized them based on their physiological and molecular analyses. Finally, we established the optimum culture conditions for the differential induction of CMCs and higher accumulation of TIAs. The cell and organ cultures of Catharanthus are being used as a means of producing important TIAs. Use of CMCs can be an important strategy for augmenting alkaloid production from plant cell cultures by avoiding the limited growth of suspension culture derived from DDCs.
Acknowledgements
This research was supported by the 2014 KU Brain Pool of Konkuk University.