

1 Introduction
The human gut microbiome can be thought of as our neglected organ [1]. It is comprised of more microbial cells than the remainder of our body, can weigh two kilograms, more than most other organs, and has a considerable metabolic activity. The microbial organ is not indispensible, as germ-free animals are viable. However, their development is impacted as their gastrointestinal tract, immune system and even brain are not fully mature, and we do not know whether their life outside of the strictly sterile conditions where they are maintained would be possible. The microbial communities that compose the neglected organ are altered in numerous diseases, in particular the chronic ones, which are constantly increasing in the industrialized societies [2]. This raises the possibility that the organ may play a role in these diseases. But why has it remained neglected, while it clearly should have been investigated? A simple answer is that there were no appropriate tools to study it. A traditional way to characterize microbial communities is to enumerate the species that they contain by culturing them in appropriate media, but for most of the microbial species from our gut we still do not have such media and do not know how to grow them reliably. The situation has changed by the advent of molecular methods precise enough to detect most of the species and determine their abundance.
2 Quantitative metagenomics for microbiome assessment
The method has been developed in the European MetaHIT consortium (http://www.metahit.eu/), operational between 2008 and 2012, and relies on the advent of new generation sequencing (NGS) techniques, capable of generating millions of sequencing reads in parallel and the numerical tools capable of handling Big Data (Fig. 1). A catalog listing the genes of intestinal microbes is central to the approach. It is used to determine the presence and the abundance of each gene in any sample under study, that is, the gene profile of any individual at a given point in time. A view of the microbiome of unprecedented precision is thus obtained. Comparisons of microbiomes (gene profiles) of different individuals, say patients and healthy controls, reveal the genes and species that distinguish them, by their presence or their abundance. The contrasting genes and species can be used, in turn, to develop powerful diagnostic and even prognostic algorithms of clinical relevance.
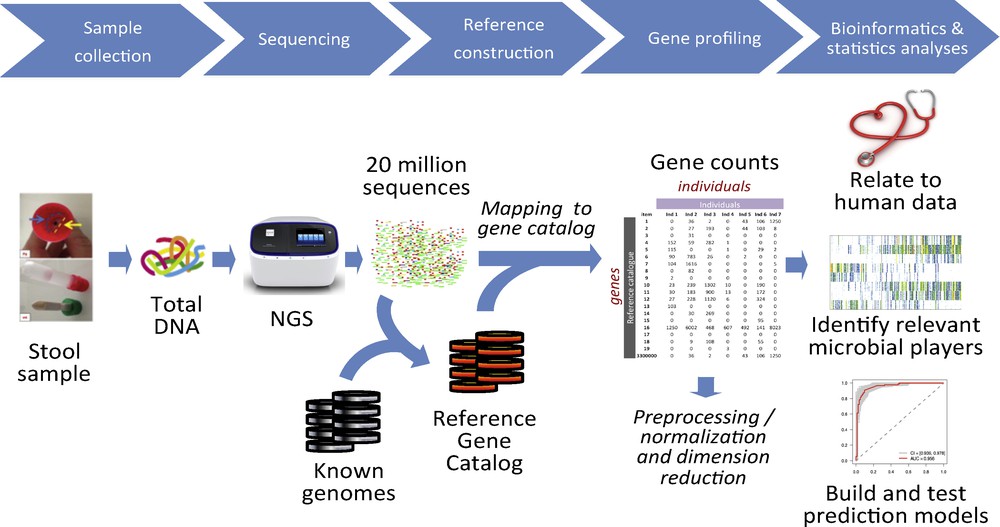
Quantitative metagenomics for the characterization of the human gut microbiome. Total DNA is extracted from a stool sample, sequenced to generate millions of reads; the reads are mapped to a reference catalog that lists all the known gut microbial genes, and a gene count table is generated for each sample. The counts are converted into gene profiles, the profiles are related to bioclinical data, and the models of clinical relevance are generated.
The first gene catalog was established in 2010, ten years after the sequencing of the human genome, by the analysis of 124 individuals of European origin [3]. It contains 3.3 million genes (> 99% bacterial, remainder of viral and eukaryote origin), 150 times more than our own genome, and was dubbed our other genome. An updated version of the catalog was released four years later. It contains 9.9 million genes, found by analyzing ten times more individuals from three continents (Europe, Asia and North America), and the sequenced reference genomes of cultured gut species [4]. Notably, the number of genes found in at least 5% of analyzed individuals increase little with each additional sample, whereas that of genes harbored by only a few individuals continue to increase, without sign of saturation. The former may approximate “core” components of the microbiome, whereas the latter can be viewed as “individualized” parts, corresponding likely, on the one hand, to transient rather than to resident gut species, introduced from the environment (e.g., microbes used in food fermentation) and, on the other hand, to strain differences between the resident species. The core part, which starts to be described in a rather comprehensive way, is most useful to address health-related questions of large populations.
Of course, genes are encoded on various genetic units, such as bacterial chromosomes or sub-chromosomal elements (phages, plasmids, CRISPR elements, virulence islands, etc.). A method to cluster genes carried on the same genetic units was developed in the MetaHIT consortium [5]. It is based on two simple facts. First, the genes carried on the same genetic unit must have the same abundance. Second, the abundance of different genetic units varies greatly between individuals (10- to 1000-fold, as assessed using known reference genomes present in more than 90% of the individuals [3]). Therefore, the genes that co-vary in abundance when different individuals are analyzed are encoded by the same genetic units.
Two types of clusters were found, differing by the number of genes. The first, containing more than 700 genes, correspond to bacterial species and are denoted MGS for MetaGenomic Species, the second centered on 50 genes and denoted MGU (for MetaGenomic Units) to smaller genetic elements. Some 740 MGS were revealed, and 85% of them were never isolated and had no closely related reference genomes, for about 240 high-quality genomes were assembled from short sequencing reads. Almost 10-fold more MGU were found; many of those were observed only in the presence of a given MGS, as expected, for instance, for a phage, which needs a specific host. The ensemble represents the most detailed species and sub-species level description of the human gut microbiome.
3 Microbiome-based diagnostics
The method, quantitative metagenomics, and the detailed description of the microbiome, based on MGS and MGU, largely set the scene to investigate the composition of the gut microbial communities in the disease. As an example of the power of the approach, likely most advanced today, we can cite that of liver cirrhosis [6].
A cohort of 123 patients and 114 healthy controls was studied and a massive gut microbial dysbiosis was detected in the former. First, patients have lost a large proportion of their microbial richness, some 25%, as is often observed in chronic diseases (Crohn's disease microbiome as an early harbinger [7]). Some 28 MGS, which were rare in healthy individuals, were overabundant in patients – collectively, they constituted a large fraction of the microbiome, up to 40%! These invading species displaced another 38 that are common and abundant in the healthy individuals. Interestingly, the invaders were mostly the species that are normally found in the mouth, but also food-borne pathogens. How could they ever become established in the gut? Likely, because the bile salt production is deficient in liver cirrhosis. Bile is toxic for most microorganisms but the normal gut residents thrive in its presence – it can be viewed as a barrier that protects the gut ecosystem from foreign microbes.
The massive dysbiosis found in liver cirrhosis allows constructing powerful diagnostic algorithms. Using only seven most informative MGS, liver cirrhosis can be detected by stool analysis, in a fully non-invasive way, with an accuracy of about 95%. Microbiome-based diagnosis has a potential to replace the current diagnostic golden standard, the invasive liver biopsy.
4 Microbiome-based patient monitoring
What could be the physio-pathological consequences of the massive invasion of the gut by oral bacteria? Quantitative metagenomics detects every gene of a catalog and allows carrying out metabolic reconstruction of the dysbiotic microbiome. It has revealed that the microbiome found in liver cirrhosis could overproduce ammoniac, a neurotransmitter gamma-aminobutyric acid (GABA) and impact manganese metabolism. These three factors are associated with a serious complication of liver cirrhosis, the hepatic encephalopathy; possibly, the altered microbiome aggravates the disease. Indeed, comparing the patients with the lowest load of the invading species with those with the highest load has revealed that the latter have significantly worse clinical scores (Model for End stage Liver Disease [MELD] and Child–Turcotte–Pugh [CTP]); their disease is more serious [6]. This example illustrates how microbiome analysis can be used to monitor the progression of the disease.
5 Microbiome-based risk detection
The detection of the risk to develop a chronic disease is a first step towards prevention, which can barely be achieved today. A study of a Danish cohort of some 292 individuals has revealed that almost one in four has lost a substantial proportion of his/her gut microbial richness, on average 40% [8]. This loss of richness was accompanied with a substantial alteration of the composition of gut microbial communities. Microbe-poor individuals had higher levels of pro-inflammatory species and lower levels of anti-inflammatory species. Consistently, the analysis of the genetic potential of the former indicated a higher propensity to synthesize the pro-inflammatory endotoxin LPS. In parallel, the capacity to synthesize butyrate, a substance necessary for the enterocytes that our own body cannot make, appeared to be significantly decreased. Most interestingly, the microbe-poor individuals displayed higher adiposity, insulin resistance, dyslipidemia, and inflammation, which, taken together, are signs of predisposition for type-2 diabetes, hepatic and cardiovascular complications and some types of cancer. Not only were these individuals predisposed to develop serious chronic diseases, but they had also, when obese, put on significantly more weight over the past 9 years than their microbe-rich counterparts, thus aggravating their health status.
A similar analysis of a French cohort of 49 individuals confirmed these observations: microbe-poor and rich individuals were found, with comparable phenotypes to those revealed in the Danish study [9]. The individuals at risk can be identified with high accuracy, close to 95%, using an algorithm based on only six informative MGS.
6 Risk alleviation by microbiome remodeling
Ideally, risk detection should be accompanied by risk alleviation. Can the lost microbiome richness be recovered? Nutritional data revealed an association of the richness with the consumption of fruit and vegetables, which could be mediated by the high fiber content of these foods [9]. An intervention study in the French cohort, with calorie-restricted diet having high fiber and protein but low fat content led to an improvement of the bioclinical parameters, as expected, but also to an increase of the richness of microbe-poor individuals, by 30% [9]. These encouraging results prompt further exploration of the possibilities to prevent chronic disease by microbiome remodeling.
7 Cause or consequence – an irrelevant question?
A question often asked is whether the gut microbiome alterations are a cause or a consequence of the disease [10]. However, a better question may be whether the altered microbiome contributes to the disease. The case can be well illustrated, once again, by liver cirrhosis. The three main causes of cirrhosis are virus infection, alcohol consumption, and obesity, which all lead to liver dysfunction; it is unlikely that microbiome alterations play a major role in triggering this disease. The resulting liver dysfunction likely promotes microbiome alteration; we hypothesize that bile production deficit renders the gut permissive to microbes normally foreign to this ecosystem. That would be a clear case of microbiome alteration as a consequence of the disease. However, the altered microbiome, in turn, may aggravate the disease, as it has the potential to overproduce toxic substances such as ammoniac and GABA, and to impact manganese metabolism; all three are thought to play a role in hepatic encephalopathy [11]. Interestingly, treatments that are currently used, laxatives, antibiotics, and enemas, actually target the microbiome. The benefits are only temporary, likely because they do not prevent the reconstitution of the harmful microbiome; a more permanent modulation could be sought as a novel way to treat this disease. We suggest that this may well be the case in many other diseases, and, whatever the trajectory that leads to the alteration of the neglected microbial organ of our body, it needs to be treated to the best of our abilities, in order to improve the patients’ health.
In a similar vein, inflammation is a facet of many chronic diseases and might promote their advent, but is not necessarily their immediate cause. The loss of gut microbiome richness found in a high proportion of otherwise healthy individuals corresponds to an alteration of the neglected organ, which promotes inflammation, likely due to endotoxin production [8]. The treatment of the altered neglected organ, aiming at recovering its richness, might reduce the risk of developing a chronic disease.
8 Ways forward
A panel of ways to modulate the microbiome can be envisaged and has indeed begun to be explored. It extends from nutritional interventions [9] all the way to organ transplantation [12], and includes treatments with molecules (promoters of beneficial components of the community or inhibitors of the potentially harmful ones, e.g., prebiotics and narrow range antibiotics, respectively) and living organisms (probiotics, artificial communities). Translating these novel ways into practices of societal impact will require industry carry-over – the field has reached the level of maturity where this is already happening, as witnessed by interest of early start-ups such as Enterome (http://www.enterome.fr/), worldwide actors such as Danone or Johnson and Johnson as well as investment funds, such as Seventure (http://www.latribune.fr/entreprises-finance/industrie/chimie-pharmacie/le-microbe-nouvelle-star-des-labos-554755.html, for a recent public media view). Hopefully, public policies concerned with health and disease will begin to reflect this ongoing rupture.
9 Conclusion
Knowledge of the human microbiome will alter our views on health and disease and likely lead to novel ways to preserve health and better treat the disease. Mechanistic insights into the interactions between the microbial organ and the remainder of our body should be vigorously pursued, as they will lead to a better understanding of our biology and lay solid ground for interventions aiming at achieving these goals. But it is safe to say that these insights will take long to be acquired. In the meantime, without wasting time, even the current, albeit limited, knowledge of the microbiome should encourage us to strive to restore or preserve health by modulating unhealthy or toxic microbiomes.
Disclosure of interest
The author declares that he has no competing interest.
Acknowledgments
The work in author's laboratory was supported, in part, by the Metagenopolis grant ANR-11-DPBS-0001.