


 CC-BY 4.0
CC-BY 4.0
1. Reconstructing the tree of life: an ambitious goal initiated by Charles Darwin
The ambitious aim of reconstructing the tree of life consists in representing all 8 million extant species [1] on a single tree that would recapitulate their evolutionary—or phylogenetic—relationships. In this tree, leaves represent extant species and nodes their common ancestors. Establishing a phylogenetic tree of life is first and foremost a way of listing and classifying the diversity of life on Earth. The tree of life is particularly valuable to conservation efforts, because it rigorously documents the diversity of living organisms and thus informs on the impacts of environmental upheavals. The distribution of endangered species along the tree of life is informative to predict which branches are the most likely to collapse and lead to the greatest losses in terms of evolutionary diversity [2].
The first sketch of a species tree is attributed to Jean-Baptiste de Lamarck, in 1809 [3]. It was only 50 years later that Charles Darwin definitively popularised the concept of the tree of life, in his seminal work The Origin of Species [4]. We also owe Ernst Haeckel many of the terms still in use in the field of evolutionary biology; inspired by Darwin’s writings, he proposed the term “phylogeny” for the first time [5]. Early phylogenetic studies used anatomical similarities between species to predict their evolutionary proximity. A century later, the DNA molecule was recognised as a “document of evolutionary history”: a powerful lever for reconstructing evolutionary relationships between species [6]. It became apparent that the mechanisms through which DNA is modified and transmitted imply that the similarity of DNA sequences between species is an indicator of their relatedness. This was followed by the advent of molecular phylogeny, and a continuous improvement of sequence evolution models, which made it possible to infer the history of life on Earth ever more completely and accurately.
Despite the advent of genomics and the refinement of DNA sequencing techniques, several nodes of the animal phylogeny remain debated within the scientific community [7, 8]. One of the most controversial nodes is the one at the base of the animal tree: which of sponges or ctenophores diverged first from the rest of animal species? This question is hotly debated, partly because its resolution will shed light on the evolution of neurons and muscles. Although morphologically distinct [9], neuronal and muscle cells are present in ctenophores and most of the rest of the animals, but are notably absent from sponges. The different phylogenies proposed at the base of the animal tree imply different evolutionary scenarios for the origin of neurons and muscles. For example, the “ctenophores-first” hypothesis implies either a common origin in the ancestor of animals with a secondary loss in sponges, or independent evolution in each of the two groups (ctenophores and the rest of the animals). Thus, resolving this node will provide the necessary framework to evaluate these evolutionary scenarios. A second challenge concerns the position of the enigmatic marine worm Xenoturbella, nicknamed the “deep-sea purple sock” because of its simplified morphology (no eye, no digestive system, no brain). The latter was alternatively placed with molluscs, as the sister group to all bilaterian animals, or close to the sea stars group. Finally, because of their impressive diversity, considerable work remains to be done to draw a complete picture of the evolution of ray-finned fishes (actinopterygians) [10]. Our work has focused on resolving a debated node at the base of the largest fish clade: the origin of teleost fishes.
2. Fifty years of debates to resolve the origin of teleost fishes
The teleost group encompasses more than 96% of all fish species. With a total of over 30,000 recorded species, it includes as many species as the Tetrapods (Amphibians, Mammals, Birds and Reptiles) (Figure 1A). Teleost fishes are subdivided into three groups: the Elopomorpha (tarpon, moray eel, eel), the Osteoglossomorpha or “bony-tongued fish” (arowana, mormyrid) and the Clupeocephala, which make up the majority of teleosts (zebrafish, tetraodon, stickleback, cod, pike, etc.). It is estimated that the last common ancestor of these three groups dates back to the Triassic period, around 250 million years ago [11].
Evolutionary relationships at the origins of teleost fishes. (A) Phylogenetic position of teleost fishes within vertebrates. Species listed to illustrate each of the three major groups (Clupeocephala, Osteoglossomorpha, Elopomorpha) correspond to species considered in our study, including those for which we generated new genomes (grey background). See the original study [12] for the full scientific names of included species. (B–D) Different topologies proposed at the base of the teleost tree: Elopocephala topology [13, 14, 15], Osteoglossocephala [11, 16] and Eloposteoglossoceephala [17, 18, 19]. Figure adapted from [12].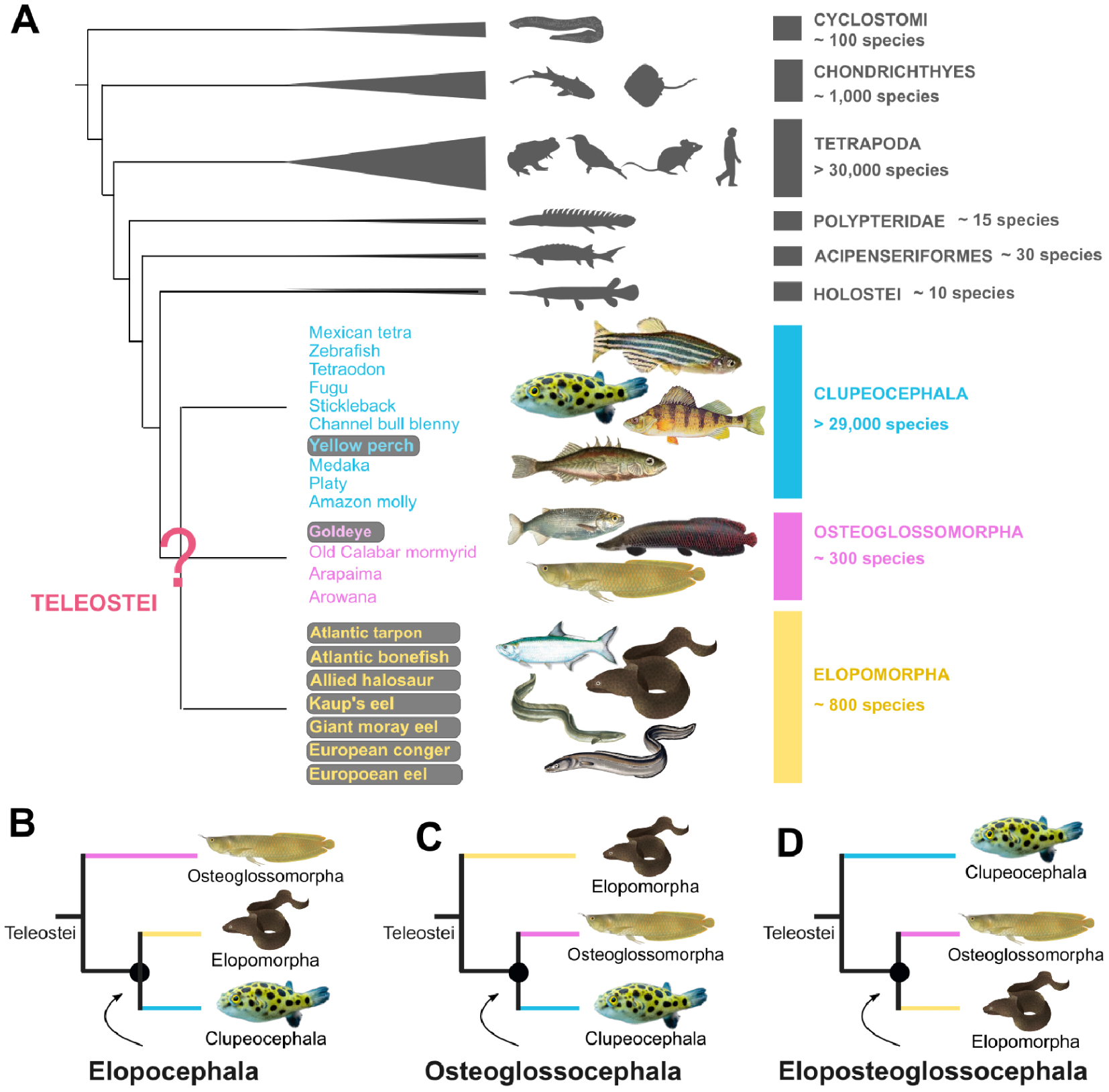
Resolving the evolutionary relationships between these three groups represents a major challenge, one which the community has been grappling with for over 50 years. The first studies were based on anatomical criteria and the analysis of fossils. They first proposed to group the Elopomorpha together with the Clupeocephala (Ref. [15]; Figure 1B), and later the Osteoglossomorpha with the Clupeocephala (Ref. [13]; Figure 1C). This question was subsequently revisited with the advent of molecular phylogeny methods which, although considering increasingly large quantities of data, have alternately supported each of the three possible groupings (Refs. [14, 17, 18, 11, 16, 19]; Figure 1B,C,D).
Several hypotheses can be put forward to explain such incongruences across studies. A first argument proposes that the instability of the Osteoglossomorpha and Elopomorpha positions in the reconstructed phylogenies might be due to their large under-representation (on average 14 times fewer species included than Clupeocephala in the previously cited studies). Other methodological considerations point to the diversity in the methods, and the relevance of technical choices made to model the evolution of sequences. Briefly, two main families of approaches are generally used: (i) the “concatenation” method, which considers all of the DNA sequences in a single block to directly reconstruct the phylogeny of species, and (ii) the “consensus” method, which considers genes separately to reconstruct a tree for each and deduce the phylogeny. The advantage of the concatenation method is that the quantity of data considered maximises the phylogenetic signal, but the disadvantage is that it is based on the assumption that all sequences share the same evolutionary history, a simplification rarely verified in practice. In contrast, gene trees of the consensus method are more prone to estimation errors due to lower statistical power, but this method accounts for distinct evolutionary histories across different parts of genomes. A phenomenon known as “incomplete lineage sorting” is most often responsible for these discordant evolutionary histories within the same genome. Incomplete lineage sorting occurs when pre-existing genetic differences within an ancestral population (different alleles of a gene, for example) are retained differentially in descendant species. For instance, if two alleles A and B of the same gene existed in the ancestral population of teleost fish and each of the three major groups randomly retained either allele A or allele B, then the evolutionary history of the sequence of this gene will not necessarily follow the phylogeny of the species. The effect of incomplete lineage sorting is all the more important when several groups of species diverge from each other in a short period of time, as was the case at the origins of teleost fishes. Introgression is another biological phenomenon that can blur the phylogenetic signal: the occurrence of hybridisation between ancient populations of the three major groups could have led to exchanges of DNA between lineages after they had separated. Consensus methods often model incomplete lineage sorting, but rarely introgression.
Finally, one of the main challenges in reconstructing the phylogenies of teleost fishes is tied to their complex evolutionary history: all extant teleosts descend from an ancestor that underwent a whole-genome duplication event [20]. As a result, many genes still exist in two copies within teleost genomes, making it difficult to identify “marker” genes, i.e. genes that are comparable between species and can be leveraged for phylogenetic analyses. Specifically, the challenge consists in distinguishing orthologous genes (genes that descend from the same copy of the ancestral gene) from paralogous genes (genes that descend from duplicated copies). The duplication event that separates paralogous genes from one another introduces a discrepancy between the evolutionary history of the gene and of the species. Orthologous gene sequences, on the other hand, directly reflect the evolutionary history of species and are therefore good markers for reconstructing their phylogeny. Although the difficulties encountered in phylogenetic reconstructions can be explained by a combination of several of the factors mentioned above, the confounding effect of genome duplication has been formally demonstrated [21] and has probably largely contributed to the incongruities reported at the origins of teleost fishes. A parallel can be drawn with the rapid diversification of the three main families of Salmonidae whose evolutionary relationships have long been ambiguous [22], linked to a whole-genome duplication event in their ancestral lineage.
3. A reanalysis in the light of new genomic data
In order to resolve the phylogenetic relationships at the origin of teleost fishes, we performed new analyses, designed to mitigate the limitations of previous studies. As part of our study, we generated new genomic resources, in particular for the Elopomorpha for which we sequenced the genomes of 7 species (Figure 1A). In total, we considered 25 genomes (Figure 1A), carefully selected to avoid significantly over-representing any one group over the others: 7 Elopomorpha, 4 Osteoglossomorpha, 10 Clupeocephala and 4 non-teleost vertebrates.
The reconstruction of a molecular phylogeny involves three main steps: (i) the identification of marker genes, present and confidently identifiable across all considered genomes (orthologous genes), (ii) the alignment of these gene sequences across species, to reveal positions that have changed during evolution (iii) the inference of a phylogenetic tree based on the observed sequence changes in the sequences. To identify marker genes across teleost fishes, a task made complicated by their ancient genome duplication event, we built upon our previous work [23, 24]. We had developed methods specifically tailored to the specificities of teleost genomes, enabling to establish a set of marker genes both more complete and more robust than leveraged in previous studies. This ortholog identification method is based on a signature left in the genomes following whole-genome duplication events. Initially, all of the chromosomes are duplicated, but, subsequently, duplicated chromosomes evolve independently and accumulate distinct gene losses and genomic rearrangements, making it possible to differentiate and identify them across species. We identify orthologous genes on the basis of their sequence conservation, but also on the conservation of their local genomic environment, which reflects their common chromosomal origin [23, 24]. Using this approach, we consider a set of 955 marker genes, representing around 5% of the complete gene repertoire, for a total size of aligned gene sequences of 2,328,657 nucleotides. In comparison, the alignments analysed in the most comprehensive previous studies [18, 11, 19] comprised between 500,000 and 1,000,000 nucleotides, i.e. approximately 2 to 5 times less.
We also took advantage of a wide range of different methodologies: we reconstructed a total of 16 phylogenetic trees, through both concatenation (direct inference of a single tree based on the entire gene set) and consensus (reconstruction of one tree per marker gene, subsequently reconciled into a single phylogeny) approaches. All these phylogenetic analyses converged to the same topology: the Eloposteoglossocephala phylogeny (Figure 1D). Although proposed by several previous studies, this is the first time that this topology has been supported regardless of the methodology employed.
4. Resolving the origins of teleost fishes through innovative methods
These new phylogenetic analyses represent an important step towards shedding light on the evolutionary relationships at the origins of teleost fishes. Nevertheless, the main contribution of our work lies in the large size of the dataset considered (number of genes), an advantage highlighted by each of the previous studies compared with their predecessors. With the aim of breaking apart from these previous debates, we also set out to take advantage of innovative molecular phylogeny methods based on novel markers.
Traditional molecular phylogeny methods leverage changes observed in the DNA sequences of genes. However, sequences are not the only genomic feature that accumulates changes over time. Genome structures are also dynamic, and involve, over the course of generations, changes in the ordering of genes along the chromosomes. These modifications accumulate more slowly than changes affecting sequences, and therefore offer a complementary perspective to study genomes evolution. Moreover, gene order evolution is potentially less affected by introgression [25]. Although the relevance of structural genomic changes for evolutionary studies was demonstrated almost a century ago [26], it is only very recently that they have started to be used to reconstruct species phylogenies [27, 12, 28, 25].
The mechanisms governing the evolution of gene organisation on chromosomes—the evolution of synteny—remain less characterised than those leading to sequence changes. As a result, there is no well-established probabilistic model to describe the evolution of synteny. In the absence of models, we applied methods that estimate evolutionary distances between pairs of genomes, and subsequently reconstruct a tree that best reflects these distances. Here, we estimated evolutionary distances by quantifying the degree of gene reordering between two genomes under consideration (Figure 2A,B), and used the Neighbor-Joining algorithm [29] to reconstruct a tree from this distance matrix (Figure 2C). The Neighbor-Joining algorithm was previously widely used in the field of molecular phylogeny, before the advent of probabilistic methods. We also applied a similar method (PhyChro, [27]), which offers an improvement on the Neighbor-Joining method, to adapt it to genome organisation markers. In particular, in the PhyChro algorithm, the distance calculation between a pair of genomes also examines all the other genomes included in the analysis, in order to specifically consider the genomic rearrangements that are the most informative to reconstruct the phylogeny.
Teleost fishes phylogeny based on gene order evolution. (A) Definition of a distance between two genomes based on the conservation of gene adjacencies. Genes are represented by rectangles along a DNA segment, with “marker” genes (i.e. genes identifiable in all considered genomes) shown in colour. A measure of similarity is calculated on the basis of the proportion of conserved adjacencies between marker genes in the two genomes being compared, then transformed into a distance. (B) Pairwise distance matrix obtained for the 25 genomes considered and 3041 marker genes, based on the d distance introduced in (A). The end-to-end distance covered by the markers considered corresponds on average to 86% of the genome. (C) Phylogenetic tree reconstructed from the distance matrix using the Neighbor-Joining algorithm [29]. Osteoglossomorpha are grouped with Elopomorpha (Eloposteoglossocephala topology, Figure 1D), with strong support (100% bootstrap).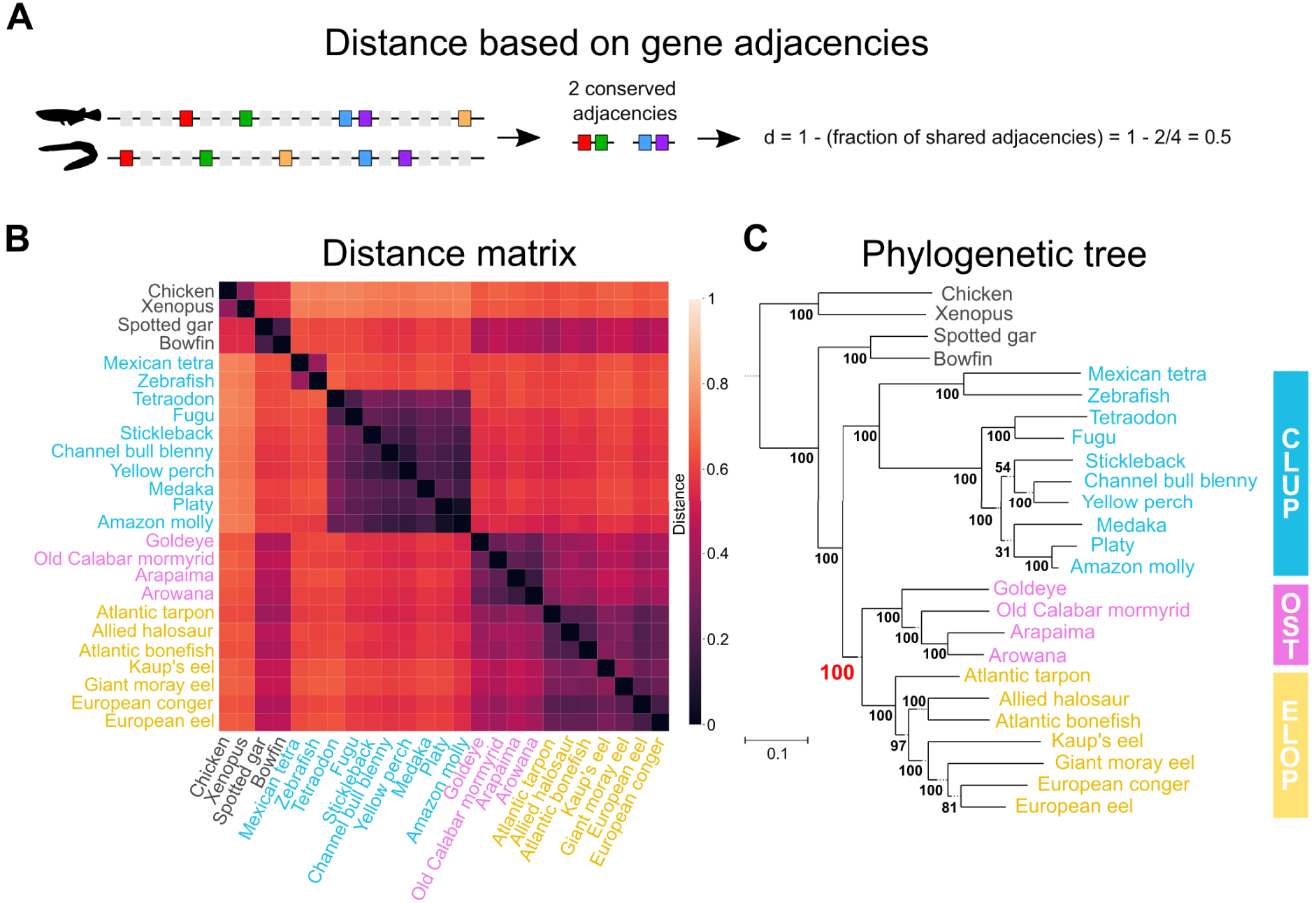
We reconstructed a total of five teleost phylogenies based on the organisation of their genomes, examined at different scales (in particular: conserved adjacencies between marker genes, ordering of gene blocks and organisation of entire chromosomes). In each of the five reconstructed phylogenies, we again recover the Eloposteoglossocephala topology, which groups the Elopomorpha with the Osteoglossomorpha (Figure 2C). We note that these different approaches use marker gene sets identified by different strategies (see [12] for more details), implying that the result is robust to the differences across considered gene sets. In conclusion, through a wide range of complementary phylogenetic analyses, we have been able to resolve the evolutionary relationships at the origin of teleost fishes and thus demonstrate that eels are more closely related to bony-tongued fishes than to the rest of the teleost fishes.
5. Towards a more systematic use of synteny to solve the tree of life
One fundamental prerequisite has to be met in order to use genome structures as a lever to trace their evolutionary relationships: the genomes considered must be of sufficient quality to provide a complete and accurate representation of gene orderings along chromosomes. These high-quality genomic resources are already available for many species, but their quantity is set to explode in the coming years, thanks in particular to many large-scale biodiversity sequencing projects (African BioGenome Project, ATLASea, Darwin Tree of Life, Earth Biogenome Project, European Reference Genome Atlas). In this context, a more systematic use of synteny to reconstruct phylogenies is becoming a feasible and promising next step, although this still presents many methodological challenges [30]. In particular, deep nodes remain intrinsically difficult to resolve due to the substantial amount of elapsed evolutionary time, which erodes phylogenetic signal both in DNA sequences and genome structures. For the biological signal provided by synteny to be usable in phylogenetic analyses, gene order must be variable between study species without being completely shuffled. Defining indicators that quantify synteny conservation and degradation limits will represent a crucial step towards defining the optimum range of application for these new methods.
Recently, a study examined synteny evolution patterns to investigate the evolutionary relationships at the origin of animals [28], a node dated around 650 million years ago. This work evidenced similarities between the organisation of the genomes of sponge and other animals, thus supporting a phylogeny in which ctenophores constitute the sister group of a clade bringing together sponges and the rest of animals. This surprising result, which contradicts the most recent molecular phylogeny analyses [8, 31, 32], is galvanising research efforts to better understand the methodological and/or biological reasons underlying these incongruences. The thousands high-quality genomes that will become available in the near future offer unprecedented opportunities to understand the mechanisms that govern the evolution of genome organisation, and re-examine many controversial branches of the tree of life.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was supported by the Agence Nationale de la Recherche, France (ANR) on the GenoFish project, 2016–2021 (grant No. ANR-16-CE12-003) and the European Union Horizon 2020 research and innovation program under Grant Agreement No 817923 (AQUA-FAANG). EP is currently supported by a Newton International Fellowship from the Royal Society (NIF/R1/222125).
Acknowledgements
We thank all of our co-authors from the original study, for their contribution to the previous work that we cover in this review article: Alexandra Louis, Jerome Montfort, Olivier Bouchez, Céline Roques, Carole Iampietro, Jerome Lluch, Adrien Castinel, Cécile Donnadieu, Thomas Desvignes, Christabel Floi Bucao, Elodie Jouanno, Ming Wen, Sahar Mejri, Ron Dirks, Hans Jansen, Christiaan Henkel, Wei-Jen Chen, Margot Zahm, Cédric Cabau, Christophe Klopp, Andrew W. Thompson, Marc Robinson-Rechavi, Ingo Braasch, Guillaume Lecointre, Julien Bobe and John H. Postlethwait.