



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Cancer remains one of the leading causes of mortality worldwide, encompassing a diverse array of pathologies characterized by dysregulation of key biological processes that regulate cellular and tissue homeostasis, such as cell division, differentiation and tissue homeostasis [1]. Although its initiation generally occurs within a specific organ or tissue, malignant cells can subsequently spread to other parts of the body and establish secondary tumors through metastasis. Known since ancient civilizations, Egyptian, Greek, Roman, as well as in traditional Chinese medicine, cancer was observed in a new light with the advent of microscopy in the 19th century, enabling, among other advances, the detection of metastases. During these early phases of research on tumorigenesis, several hypotheses were proposed [2]. Theodor Boveri is credited with the first formulation of the widely accepted theory on the origin of cancers, known as the “somatic mutation theory” [3]. This theory posits that cancer arises from a chromosomal alteration, promoting the transformation of a normal cell into a malignant state, subsequently inherited by its daughter cells.
In addition to DNA mutations, epigenetic alterations also play a crucial role in cancer. Epigenetics refers to the study of mechanisms and molecules involved in the inheritance of different gene expression profiles from the same DNA sequence [4]. These regulatory mechanisms are essential not only during development and adulthood but also in aging and in most human pathologies, including cancer [5]. Although epigenetics is often considered of therapeutic interest due to its role in tumor progression and metastasis [6, 7], recent data, which will be discussed in this article, suggest that epigenetic alterations can also serve as drivers of tumorigenesis. These discoveries prompt a reconsideration of the role of the DNA sequence in cancer etiology and call for a rethinking of our cancer prevention and treatment strategies.
2. The somatic mutation theory and its impact on modern oncology
In its first formulation in 1914 [3], Theodor Boveri postulated that cancer could originate from chromosomal abnormalities, particularly defects in chromosome segregation during cell division. This hypothesis found early support with the discovery of abnormal chromosomes in leukemias [8, 9] and was further substantiated by the identification of the first oncogenes by several laboratoires [10]. These discoveries were integrated into a modern version of the “somatic mutation theory” (SMT) to suggest that cancer arises from genetic mutations. However, it is important to emphasize that Boveri did not focus solely on DNA. Chromosomes also consist of associated proteins and RNAs that play a crucial role in regulating gene expression and maintaining chromosomal integrity. Beginning in the 1980s, extensive research into oncogenes and tumor suppressor genes reinforced a mutation-centric view of cancer. This led to the development of a comprehensive catalog of oncogenes and tumor suppressors, establishing a molecular framework that currently shapes cancer research and therapeutic development.
In a seminal article published in 1976 [11], Peter Nowell hypothesized that cancer development occurs in multiple stages. An initial cell (or a small group of cells) undergoes a primary alteration that makes it neoplastic, granting it a proliferative advantage. Subsequent alterations, primarily driven by mutations, would promote clonal selection, ultimately leading to the formation of malignant tumors. This publication is a cornerstone in oncogenesis as it introduces the concept of tumor-initiating cells and their clonal evolution. Following the discovery of oncogenes and tumor suppressor genes, a merger occurred between the hypothesis of clonal initiation of tumorigenesis and the somatic mutation theory, leading to the proposition that mutations arise in the cancer-initiating cell(s) (Figure 1). Alternative hypotheses suggesting a non-genetic origin of cancer, centered on dysregulation of gene regulation, were largely dismissed. The notion that tumors contain clones of cells that have dominated through “evolutionary competition” further strengthened the scientific community’s belief that large-scale sequencing of tumor genomes would uncover all cancer-relevant genes.
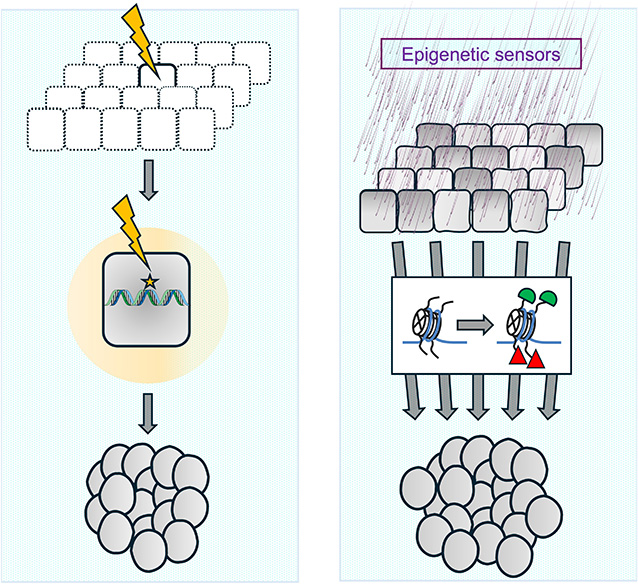
Mutational versus epigenetic origin of cancer. On the left, the dominant hypothesis on the origin of cancers, based on the somatic mutation theory, is depicted. According to this theory, stochastic mutations, potentially caused by exposure to mutagenic agents (illustrated by a yellow arrow at the top), can sometimes activate oncogenes or inactivate tumor suppressor genes, thereby generating the tumor-initiating cell or “cancer stem cell” (center). Subsequent mutagenic events and the selection of specific cell clones ultimately lead to the development of aggressive cancer (bottom). On the right, the hypothesis of the epigenetic origin of cancers is shown. Exposure to non-mutagenic carcinogens, nutritional and metabolic changes, or other sources of stress (top) could affect a group of cells, altering the chromatin state of some of their genes (center, with altered epigenetic modifications represented in green and red). This induces changes in gene expression, which, if self-sustaining, could initiate a tumorigenesis process that persists even after the stimulus that caused the initial epigenetic alteration has ceased. Masquer
Mutational versus epigenetic origin of cancer. On the left, the dominant hypothesis on the origin of cancers, based on the somatic mutation theory, is depicted. According to this theory, stochastic mutations, potentially caused by exposure to mutagenic agents (illustrated by ... Lire la suite
Much like the distinction between Darwinism and neo-Darwinism in evolutionary biology (Box 1), the modern formulation of the SMT can therefore be considered a “neo-Boverian” perspective, emphasizing DNA mutations as the drivers of cancer while disregarding the potential causal roles of other alterations, such as the composition or structure of chromosomes.
Box 1
Darwinism refers to the theory of evolution of species through natural selection, as developed by Charles Darwin in On the Origin of Species [12]. This theory is based on the observation of phenotypic variations among species, particularly within closely related species, suggesting that phenotypic traits evolve over generations. According to Darwinian theory, traits are transmitted through a mechanism called pangenesis, where the entire organism contributes to heredity, notably through the budding of gemmules from its cells, especially in the reproductive organs.
Neo-Darwinism stems from the modern synthesis of the evolutionary theory of natural selection. This updated version of Darwin’s theory incorporates later genetic discoveries, particularly Mendelian inheritance and population genetics, as the foundation of evolution [13, 14]. In this modern synthesis of evolutionary theory, the source of variations among individuals of the same species lies in the different genetic information transmitted through gametes. Generally, this information is considered to reside in the DNA sequence, and genomic variations are increased by random mechanisms such as mutations and meiotic recombination. The mechanisms of selection depend on the ability of each individual to survive and reproduce within populations of each species. This ability depends on that of other individuals, on the size of populations and on environmental conditions. Epigenetics shows that information other than what is present in the DNA sequence can contribute to phenotypic characteristics. Its contribution to evolution is currently a topic of discussion within the scientific community.
With the advent of high-throughput sequencing technologies, large-scale sequencing projects involving cohorts of various cancer types were initiated. These projects rapidly identified genes frequently subject to mutations, amplifications, or deletions in cancer samples [15, 16]. Concurrently, functional studies demonstrated that these same genetic alterations can induce tumor formation in mice [17]. This body of experimental evidence, supported by epidemiological data, confirmed DNA sequence mutations as key oncogenic events. As a result, they are regularly used as biomarkers for diagnosis, prognosis, and therapeutic decision-making in clinical practice.
3. Alternative hypotheses concerning the origin of cancers
The SMT, associated with the hypothesis of cancer initiation by Tumor Initiating Cells (TICs), has become the dominant theory in oncology. However, it is important to recognize that Peter Nowell’s original formulation did not attribute the first neoplastic event to genetic mutations. In the original article, Nowell pointed out that
“The biological consequences of the primary alteration may be illustrated with various examples …The specific gene products that produce these biological consequences remain uncertain. Equally obscure is the specific genetic event which produces them. Absence of new gene products in tumor cells and the reversibility of transformation in certain culture systems has led some investigators to suggest that initiation usually involves altered gene expression rather than structural mutation. It is certainly clear that visible alterations in chromosome structure are not essential to the initial change. Transformation can take place in tissue culture and certain tumors can develop in vivo without detectable cytogenetic abnormalities”. [11]
The phenomenon of tumor reversion observed in several circumstances presents a compelling argument suggesting that mutations (whose probability of reversion is extremely low) are not causal, at least in these specific cases [18]. Furthermore, large-scale sequencing studies have revealed the limitations of sequencing. First, many mutations identified in such studies are not necessarily sufficient to explain cell transformation. These same mutations found in cancers are frequently found in normal tissues. Notably, cells harboring so-called “driver” mutations, which are thought to trigger tumorigenesis, are also sometimes present and abundant in healthy tissues [19]. Moreover, the mutation rate in normal cells is comparable to that measured in several cancer types [19]. Finally, even in cases involving oncogenes strongly associated with the emergence of cancers, such as the Ras oncogene, mutation alone rarely triggers tumor formation. Instead, it is tissue lesions that strongly stimulate tumorigenesis by modifying chromatin states and the regulation of gene expression [17].
The inability to fully account for the origin of all tumors by mutations in oncogenes or tumor suppressor genes has spurred the development of alternative hypotheses. A radically different hypothesis from SMT is the “Tissue Organization Field Theory or TOFT” [20, 21]. According to this theory, cancers do not necessarily arise from clonal or mutational events. Rather, they result from chronic disruptions in the interactions between different cellular components of a given morphogenic field within a tissue. These perturbations may be caused by exposure to carcinogens or physiological stressors, inducing stable changes in gene expression mediated by epigenetic alterations. This theory echoes the hypothesis that cells, individually or in groups, can undergo stable changes in their destiny, adopting alternative functional states without requiring genetic mutations. This process resembles the dynamic “valleys” of Waddington’s epigenetic landscape (Figure 2 and Box 2). However, unlike the stable destinies observed in normal cells, these altered states result from stochastic events or external disturbances such as exposure to carcinogens, which drive tissue-level dysregulation [22, 23]. A third hypothesis about the origin of cancers is the “evolutionary reversion” theory, which suggests that cancer cells reach a state of uncontrolled proliferation by reverting to ancestral cellular states resembling those of unicellular organisms, whose default state is proliferation. The mechanisms of proliferative inhibition, typical of tissues subject to size control, are thus lost in cancer cells [24], resulting in a stable proliferative state, at the origin of cancers.
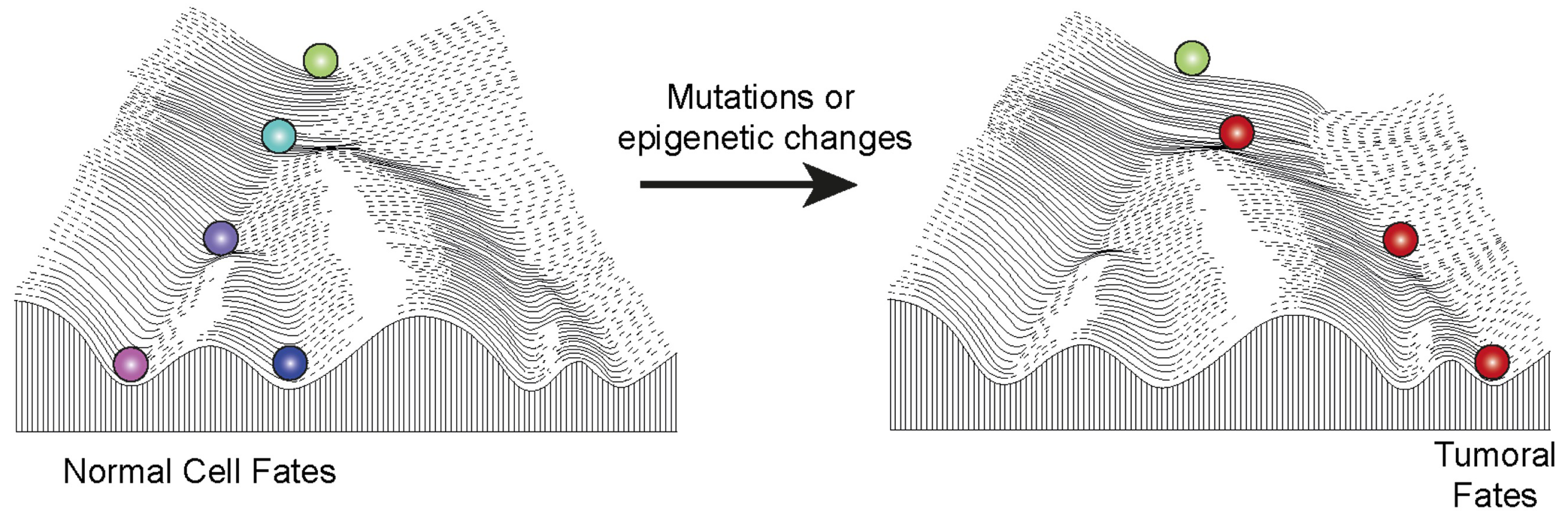
The epigenetic landscape in normal development and in cancer. The illustration depicts the famous Waddington landscape, where a marble rolls down a slope with multiple possible trajectories, determined by the hills and valleys encountered along its path. This landscape symbolizes the various cellular fates that can arise from a pluripotent cell, such as the zygote, supporting the hypothesis that epigenetic inheritance contributes to the stable transmission of cellular fates. Epigenetic components or environmental exposures contribute to shaping the landscape and lead to a variety of cellular fates. In the context of normal development (left), cells move down the hill during differentiation to acquire normal fates. However, when epigenetic components are disrupted, the landscape itself is altered (right), causing cells to take an aberrant path, which can ultimately lead to cancer. Masquer
The epigenetic landscape in normal development and in cancer. The illustration depicts the famous Waddington landscape, where a marble rolls down a slope with multiple possible trajectories, determined by the hills and valleys encountered along its path. This landscape symbolizes ... Lire la suite
Finally, the developmental constraints model, based on extensive analysis of single-cell transcriptomic data from various types of cancers in comparison to healthy tissues, suggests that the tissues of origin impose differentiation constraints on cancer cells. According to this model, cancer cells are limited in the range of cellular states they can adopt, and these constraints are determined by their tissue of origin [25]. This perspective is of considerable interest because it provides an explanation for why each tissue or cell type gives rise to a limited spectrum of cancer subtypes.
Box 2: The Waddington epigenetic landscape
The famous Waddington landscape (Figure 2) depicts a marble rolling down a slope, which can follow different trajectories depending on the valleys and hills it encounters along the way. This metaphorical illustration represents the various cellular fates that a cell, initially represented by the zygote, can adopt during its development. This landscape is commonly used to visually explain how epigenetic mechanisms contribute to the stable transmission of cellular fates once they are established by intrinsic and extrinsic signals.
Polycomb complexes can play a major role in shaping this landscape due to their ability to regulate epigenetic inheritance. They enable cells to follow specific trajectories, establishing and stabilizing different differentiated states during normal development. However, mutations or disruptions affecting the activity level of Polycomb complexes can alter this landscape. When these disruptions are strong enough, they can reshape the landscape in such a way that forces cells to follow aberrant, yet intrinsically stable, trajectories, thereby promoting the formation of cancers (Figure 2).
4. Epigenetic components and cancer
Although the precise role of DNA mutations and epigenetic changes at different stages for each cancer type is yet to be clarified, it is important to consider that factors involved in epigenetic inheritance contribute significantly to tumorigenesis [26, 27, 28]. These factors can be broadly classified into four major molecular categories.
DNA methylation primarily modifies cytosines in mammals. The maintenance of this mark over time and through cell divisions relies on the DNA methyltransferase DNMT1 and its partner UHRF1, which enhances DNMT1 activity [29, 30]. This molecular complex exhibits a unique ability to bind to the epigenetic mark 5-methylcytosine in a CG-rich DNA sequence context (mCpG) when the cytosine is hemimethylated. This occurs notably after the replication of fully methylated sequences, as during replication, each strand of DNA, which carries methylated cytosines, is paired with a new strand bearing “naive” and therefore unmethylated cytosines. The binding of the DNMT1/UHRF1 complex, followed by the catalysis of the methylation of the naive cytosine, restores the fully methylated state, thereby preserving the epigenetic memory of this mark [31]. However, DNA methylation can be modified, particularly through oxidation reactions mediated by specific enzymes called “Ten-Eleven Translocation” (TET), whose alterations are also associated with cancer [32].
Non-coding RNAs (ncRNAs) encompass several classes, each defined by its own mechanisms of production, metabolism, and specific biological functions, frequently linked to tumorigenesis [33, 34, 35, 36]. Some regulate post-transcriptional processes, like microRNAs, while others influence the transcriptional regulation of the genome. These RNAs vary in size: some are small (<30 nucleotides), while others are large (200 nucleotides or more, sometimes several hundred thousand nucleotides). Depending on molecular or cellular context, ncRNAs can play either activating or repressing roles. They can also affect other epigenetic processes such as DNA methylation or regulators of chromatin composition and architecture [37, 38, 39].
Heterochromatin is a compact and transcriptionally repressed form of chromatin [40, 41, 42] that contains many transcription-repressing proteins, as well as a large number of repetitive DNA elements. It can form chromosomal domains spanning several megabases, covered by a specific trimethylation mark of histone H3K9 (H3K9me3), which is deposited by the enzymes SUV39H1 and SETDB1. This mark is recognized by heterochromatin-associated proteins, whose binding stimulates their catalytic activity. The H3K9me3 mark also recruits the HP1 protein, which can link adjacent nucleosomes. Thus, the components of heterochromatin can both deposit and bind to the H3K9me3 mark, contributing to the compaction of their target chromatin [43]. Additionally, the presence of factors in the same protein complex that can both deposit and recognize a mark can, on one hand, stimulate the propagation of these marks to form large chromatin domains, and on the other hand, contribute to the stability of these domains and their hereditary transmission [44]. Furthermore, heterochromatin factors collaborate with other chromatin factors to transmit epigenetic inheritance across generations [43].
Finally, Polycomb proteins mainly group into two classes of complexes: PRC2 and PRC1, responsible for establishing the H3K27me3 and H2AK119Ub marks, via their EZH2 and RING1A/1B catalytic subunits, respectively [45]. Their recruitment to specific genomic regions can occur through DNA-binding proteins or ncRNAs. Similar to heterochromatin, Polycomb complexes can bind to the histone marks they deposit, therefore facilitating the transmission of epigenetic memory across cell generations and meiosis [46, 47, 48]. Their action is also reversible, notably through activator proteins capable of displacing Polycomb complexes from the chromatin (such as SWI/SNF proteins) or replacing repressive histone marks with activating marks (such as proteins from COMPASS complexes). These Polycomb factors, along with SWI/SNF and COMPASS, are often deregulated or mutated and contribute to tumorigenesis in many types of cancer [49, 50].
Over the past two decades, numerous studies have highlighted that these different epigenetic factors—both alone and in combination with other cellular components and environmental factors—play a major role in the mechanisms of tumorigenesis [5, 51, 52]. Nevertheless, the prevailing view remains that genetic mutations are paramount to trigger tumorigenesis, while epigenetics only comes into play after sequence modifications to accompany or exacerbate cancer progression.
5. Epigenetic initiation of tumorigenesis in Drosophila
Epigenetic factors being essential for the regulation of the expression of most genes, it is relevant to explore whether epigenetic changes alone can initiate a tumoral state (Figures 1 and 2). However, since epigenetic changes are often reversible, the question arises whether they can induce aberrant cellular states that are stable enough to lead to pathologies such as cancer. Genomic and epigenomic analyses of cancers do not provide a conclusive answer to this question, as they are conducted on tumors at relatively advanced stages and generally reveal the presence of mutations as well as epigenetic changes, with significant heterogeneity within tumor cells. This complexity makes it challenging to identify, among all these variations, the specific changes that initiated the first cancerous cell.
To distinguish the epigenetic contributions from genetic mutations, it is necessary to induce a purely epigenetic alteration without modifying the DNA sequence and to test whether this alteration is sufficient to initiate a tumoral process. For such a study to be conclusive, it must rule out the possibility that simultaneously occurring genetic changes are responsible for the cancer. Since mutations occur frequently, with one or more mutations arising during each cell division, it is necessary to sequence and analyze the tumors thus generated. An ideal experimental system should enable the rapid and reproducible induction of tumors in order to minimize the likelihood of genetic contributions.
Such a system has recently been developed in Drosophila melanogaster [53], a model organism commonly used for studying the fundamental mechanisms of cellular transformation. This model benefits from a wealth of knowledge and genetic tools, as well as the evolutionary conservation of mechanisms involved in tumor initiation and progression, including numerous oncogenes and tumor suppressor genes [54, 55, 56, 57].
In Drosophila, Polycomb proteins are tumor suppressors that exert their function by inhibiting the Notch, JNK, and JAK-STAT signaling pathways [49, 58, 59, 60]. Research has shown that aggressive tumors develop following depletion of Polycomb factors during larval development [61]. This system was recently adapted to test the effect of transient depletion of these proteins. Drosophila offers precise spatiotemporal control over gene expression through temperature-sensitive RNA interference (RNAi) systems. At a so-called permissive temperature, the protein of interest is present at normal levels. Raising the temperature to 29 °C activates the RNA interference system, depleting the targeted protein. At the desired time, returning the flies to the permissive temperature restores normal expression of the previously depleted factor.
Surprisingly, aggressive tumors develop after just 24 h of transient depletion of a Polycomb protein [53]. Genome sequencing of these tumors confirmed the absence of oncogenic mutations, proving that tumorigenesis is indeed initiated by the transient reduction of a Polycomb factor. This means that flies whose genome does not induce tumors at the permissive temperature develop tumors simply due to a transient temperature shift. Importantly, this transient change does not induce tumors in control Drosophila lacking the temperature-dependent depletion system. Thus, it can be excluded that the temperature change itself causes tumorigenesis. The origin of these tumors is therefore purely epigenetic.
The study of the mechanisms involved in this epigenetic tumorigenesis showed that the depletion of Polycomb proteins leads to the deregulation of a large number of genes. Some of these genes are direct targets of Polycomb proteins that bind to their regulatory regions. This binding induces chromatin condensation, thereby repressing these genes. Upon the loss of Polycomb, the transcription of these genes is activated. Once Polycomb proteins are restored to normal levels, they generally rebind to their target regions. However, the chromatin of certain genes can no longer condense properly, and a subset of genes continues to be abnormally expressed, leading to tumorigenesis (Figure 1). The transient loss of Polycomb factors therefore leads to an irreversible aberrant state of gene expression that is self-maintaining even after the return of Polycomb factors, allowing the cells to become cancerous [53].
6. Epigenetic initiation of tumorigenesis in mammals
If epigenetic factors can be the origin of cancers in flies, what about mammals? “Proof-of-concept” studies have been conducted in mice using models of multiple myeloma [62] and B-cell lymphoma [63]. In these studies, transient expression of an oncogene (MafB for multiple myeloma and Bcl6 for B-cell lymphoma) in hematopoietic stem cells led to the appearance of tumors within a few weeks, with characteristics closely resembling those of corresponding human tumors. Interestingly, even after oncogene expression ceases, tumor cells retain a stable tumor-specific transcriptional program. Moreover, the tumor cells display DNA methylation changes. In the case of B-cell lymphoma, the regions with methylation defects are enriched with motifs recognized by the oncogene Bcl6, indicating that the tumor cells have developed a stable epigenetic program following the transient oncogenic expression.
All of these data suggest that epigenetic mechanisms can initiate tumorigenesis not only in Drosophila but also in mammals. It is important to note that these proof-of-concept studies use triggering mechanisms based on transient actions (also defined as “hit-and-run”) that are designed in the laboratory. However, “hit-and-run” events, based on the principle of transient modifications to genome function, such as viral infections, can also trigger tumors [64, 65], further suggesting that natural events may also induce epigenetically driven tumorigenesis.
7. Future and biomedical perspectives on the role of epigenetics in cancer initiation
Although experimental work on epigenetically driven cancers provides a clear demonstration of the possibility that tumors can emerge without mutations, several critical points remain to be clarified. First, the precise molecular mechanisms by which normal cells undergo malignant transformation still need to be described. This is true both for studies in mice and in Drosophila. A detailed longitudinal follow-up could provide significant insights into these processes. Second, it remains unclear whether tumor cells emerge from all cells that underwent a transient disruption or if specific cell clones emerge as a result of this disruption. In-depth single-cell analyses, as well as genomic sequencing to identify potential mutations in tumor cell clones, could help understand whether genetic changes accompany or even promote the epigenetic action in triggering tumors, particularly in mice.
There are human tumors for which, despite extensive sequencing, no oncogenic mutations have been identified. These include a subset of gastrointestinal stromal tumors (GISTs) [66, 67], as well as infantile hemangiomas [68]. Another example, which has been studied in detail, is the posterior fossa ependymoma (PFA) [69], a pediatric brain cancer. In PFA, DNA methylation defects, a reduction in the activity of the Polycomb PRC2 complex proteins [70] and alterations in the three-dimensional folding of the genome have been observed in the associated genes [71, 72]. Sequencing of these tumors is often limited to the exome. When whole genome sequences are obtained, they are limited to a small number of patients and use short-read sequencing techniques that cannot analyze the repeated elements of the genome. In the future, more extensive sequencing, particularly using “long-read” techniques that allow for the sequencing of several kilobases of DNA, should help better understand if DNA mutations are present in these tumors. However, the available data suggest that, for these types of cancer, dysregulation of epigenetic factors could play a triggering role. Furthermore, even when mutations are detected, they may result from the selection of specific cell clones that emerged after the initial cellular transformation. The first tumor cells could therefore be generated by epigenetic changes but be eliminated during tumor development at a later stage. It is therefore important to continue studies using experimental models to deepen our understanding of the underlying molecular phenomena, as well as to focus on human cancers with few or no oncogenic mutations, which represent promising candidates for epigenetic cancers.
In addition to its significance for the fundamental understanding of cancer biology, this research could have implications for therapeutic approaches to certain types of cancers. In particular, therapies targeting epigenetic factors are beginning to be implemented. These treatments could play an increasingly significant role as new molecules are developed to more selectively target specific epigenetic components [73, 74, 75].
A recent epidemiological study clearly showed a significant increase in the incidence of cancers diagnosed early (before the age of 50) [76]. This study identified risk factors such as diet, alcohol consumption and tobacco use as the factors most correlated with this increase [76]. These factors are well known to induce epigenetic modifications, so it is conceivable that, rather than a massive increase in mutations, it is the alteration of cellular and tissue functions, partly due to epigenetic changes, that may be responsible for the rise in early cancer incidence.
Together, these findings highlight the need to decipher not only the mechanisms of cancer progression and metastasis but also those of its initiation. To achieve this, it is essential to establish the complete chain of causes and consequences that ultimately explains the evolution of the disease in each patient. This comprehensive understanding requires an interdisciplinary approach, encompassing fields as diverse as mathematics, social sciences and the humanities [77]. Such an approach could not only improve diagnostic and therapeutic strategies but also help develop prevention strategies based on non-mutagenic molecular mechanisms of tumorigenesis [78, 79].
Glossary
| DNMT1 | DNA methyltransferase 1 |
| UHRF1 | Ubiquitin-like, containing PHD and RING finger domains, 1 |
| mCpG | methyl-CpG |
| SUV39 | SUV39H1 histone lysine methyltransferase |
| SETDB1 | SET Domain Bifurcated Histone Lysine Methyltransferase 1 |
| HP1 | Heterochromatin protein 1 |
| PRC2 | Polycomb Repressive Complex 2 |
| PRC1 | Polycomb Repressive Complex 1 |
| EZH2 | Enhancer of Zeste Homolog 2 |
| RING1A/1B | Ring finger protein 1A/1B |
| H3K9me3 | Trimethylation of histone H3 on lysine 9 |
| H3K27me3 | Trimethylation of histone H3 on lysine 27 |
| H2AK119Ub | Monoubiquitinated lysine 119 of histone H2A |
| SWI/SNF | SWItch/Sucrose Non-Fermentable |
| COMPASS | COMplex of Proteins ASsociated with Set1 |
| JNK | Jun N-terminal Kinase |
| JAK-STAT | JAnus Kinase-Signal Transducer and Activator of Transcription |
| MafB | V-maf musculoaponeurotic fibrosarcoma oncogene homolog B |
| Bcl6 | B-cell lymphoma 6 |
| GIST | GastroIntestinal Stromal Tumors |
| PFA Ependymoma | Posterior Fossa type A Ependymoma |
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Vous devez vous connecter pour continuer.
S'authentifier