

Abridged English version
Tumor cells sequentially acquire self-sufficiency properties compared to their normal counterparts. The acquisition of these properties correlates with alterations of pro- (oncogenes, growth factors receptors, positive regulators of the cell cycle ...) or anti- (tumor suppressors, negative regulators of the cell cycle, factors controlling the genome integrity, pro-apototic factors ...) tumorigenic factors expression and/or function. These alterations can affect the mechanisms involved in the control of the DNA integrity as well as those regulating the gene expression products, i.e. the messenger RNA and the protein itself.
The DNA
The DNA can be the target of two main alterations: (i) conformational changes resulting from epigenetic modifications (methylations, aberrant post-translational modifications of histones) that disturb the compaction of chromatin and thus the accessibility of transcription factors and of the transcription machinery; and (ii) nucleotidic alterations that may lead to point mutations up to significant chromosomal rearrangements.
Conformational changes
The DNA methylation is a covalent modification exerted by DNA methyltransferases (DNMT) on cytosines of CpG di-nucleotides concentrated in short regions called CpG islands, and preferentially present within the promoter and the first exon of more than 60% of the human genes. A poor methylation of the DNA is usually observed in tumor cells and can participate in the activation of genes involved in the process of invasion and metastasis such as the PLAU (urokinase-type plasminogen activator) protease or in the promotion of cell proliferation by affecting, for example, the IGF2 (Insulin-like growth factor) encoding gene as observed in Wilm's tumors. This hypomethylation, however, mainly affects euchromatic centromeric regions or LINE (long interspersed nuclear element) repeated sequences and is suspected to be involved in the cancer associated genomic instability. A hypermethylation can also be observed within specific loci and the increased expression of the DNMT1 and DNMT3B methylases in several types of cancers correlates with a decreased in the expression of some tumor suppressors such as p16INK4A, CDKN2A and 2B (cyclin-dependent kinase inhibitor), E-cadherin, MLH1 (human mismatch repair), RB1 (retinoblastoma) or BRCA1.
Post-translational changes of histones H3 and H4 also alter the chromatin structure and a key role of HDAC (histone deacetylase) in tumorigenesis has been highlighted by the reexpression of tumor suppressor genes such as p21 as well as by a tumor regression upon an inactivation of these enzymes.
Nucleotidic changes
The cancer cells genome seems to be particularly unstable, and the aneuploidy found in most cancers might be the starting point of cell transformation before the accumulation of subsequent mutations. The three main instabilities are the NIN, the MIN and the CIN.
The NIN or nucleotide instability is rare in cancers, is caused by defects in the mechanisms of DNA repair such as NER (nucleotide excision repair) and BER (base excision repair) and leads to subtle mutations in the DNA sequence (substitutions, deletions, insertions of a few nucleotides).
The MIN or microsatellite instability, also rare in cancers, occurs at repeated genomic sequences such as polyA or polyCA, is caused by defects in the MMR (mismatch repair) system and has been associated, for example, with the development of hereditary HNPCC (hereditary non-polyposis) colorectal cancers.
The CIN or chromosomal instability seems more frequent in cancers and is suspected to result from defects in the mechanisms involved in the chromosomes condensation, telomeres stability, chromosomes–microtubules interactions or cell cycle progression (check-points). The resulting rearrangements are very heterogeneous even within the same type of cancer and can be classified into four groups: (i) the aneuploidy, an abnormal number of chromosomes that results from a loss or a gain of up to entire chromosomes; (ii) the amplification of genes involved in growth (ERBB2, EGFR ...), proliferation (c-Myc, N-Myc, K-Ras, Cycline D1 ...), apoptosis inhibition (Bcl2, HDM2 ...), drug resistance (MDR1, MPR ...): (iii) the loss of genes or LOH (loss of heterozygosity), that affects tumor suppressor genes such as those encoding p16 (CDKN2A), p15 (CDKN2B), p53, BRCA1 (Breast Cancer 1) and its counterpart BRCA2; and (iv) the gene rearrangements that are due to chromosomal translocation and lead to the expression of chimeric proteins such as the BCR-ABL fusion protein and/or that put the gene under the control of another promoter. This is for example the case of the Myc oncogene put under the control of the immunoglobulin H (IgH) heavy chain upon the t8, 14 translocation observed in some leukemias and lymphomas, in particular, the Burkitt lymphoma.
The messenger RNA (mRNA)
mRNA synthesis (transcription), maturation and stability are also steps that can all be altered during tumorigenesis.
Transcription
Transcription is a subtly regulated mechanism that can be deregulated at the DNA level by epigenetic changes or genetic alterations, but nearly all signalling pathways altered in cancers also lead to the deregulation of transcription factors (or their direct or indirect targets) expression and/or activity. Moreover many pro- or anti-tumorigenic genes encode transcription factors or proteins regulating transcription factors whose deregulation leads to the acquisition of a cellular transformed phenotype. This is particularly true for Myc, pRb/E2F or p53.
The mRNA maturation
The mRNA maturation by alternative splicing increases the number and the diversity of proteins from a unique gene. Several cis-regulatory elements are involved in this process: the splice sites themselves (GT in 5′ and AG in 3′), and some strongly degenerated sequences referred to “enhancers” or “silencers” and recognized by trans-regulatory elements such as SRP (serine arginine rich proteins) or hnRNP (heterogenous nuclear ribo-nucleo-proteins) respectively. Cancer cells exhibit frequent aberrant splicing leading to accumulated natural variants or even to the presence of cancer-specific variants that do not exist in the healthy tissue. These can be due to the appearance of mutated splicing sites such as in the case of APC, BRCA1, and CD44, but also to the deregulation of the expression of trans-regulatory elements such as SRP40 for example.
The mRNA stability
The mRNA stability is dependent on its 3′UTR region (UnTranslated Region). This region is not read by the ribosomes and contains enriched adenines and uridines specific sequences called ARE (AU-Rich Element) able to interact with trans-regulating actors such as the ARE Binding Proteins (ARE-BP). ARE containing mRNA are targeted to the exosome, a multiprotein complex exhibiting an exonucleotidic activity responsible for their degradation. Alterations of the messengers stability may result from either mutations in the ARE as reported for c-myc and cyclin D1, or from the deregulation of ARE-BP such as the ubiquitously expressed HuR whose over-expression has been observed in several types of cancers and associated with an increased expression of the c-Fos and c-Myc oncogenes, cyclin A, B1 and D1, VEGF and EGF, cyclo-oxygenase COX2 as well as the metalloprotease MMP9. MicroRNA (or miRNA) are also important regulatory elements in the control of the mRNA stability. They preferentially bind to the 3′UTR region of their mRNA targets, suppress their translation and/or induce their degradation. Many miRNA encoding sequences are located in cancer associated chromosomal loci close to oncogenes, tumor suppressors, or sites sensitive to genetic instabilities that could explain the changes in their expression level observed in cancer cells. Moreover, some miRNA themselves exhibit pro- or anti-tumorigenic properties such as MiR21 or Let7 targeting the PTEN or Ras encoding mRNA respectively.
The protein
The protein is the final product of the gene expression and the mechanisms involved in the control of its own synthesis (translation) and stability may also be altered in tumors.
Translation
The protein synthesis consists of three steps (initiation, elongation and termination) each requiring its own regulators: eIFs (eukaryotic initiation factors), eEFs (eukaryotic elongation factors) and termination factors (release factors). However, even though the eIF4E, eIF4G, eIF4A and eIF2 factors appear to be up-regulated in many tumors, and lead to an over-expression of cancer associated genes, an overall increased translation is rarely observed in cancers. Indeed, the 5′UTR of the majority of mRNAs is short (less than 200 bases), not often subject to secondary structures and thus less dependent on the helicase activity of the initiation complex. However some mRNAs encoding oncogenic factors, growth factors and regulators of apoptosis exhibit a longer and often GC rich 5′UTR region more susceptible to the formation of secondary structures and thus more dependent on the helicase activity of the initiation complex. This is particularly true for the ornithin decarboxylase (ODC), FGF2, VEGF, and cyclin D1 encoding genes.
The protein stability and degradation
The protein stability and degradation are the last steps able to maintain normal cellular protein contents. Beside post-translational changes (such as phosphorylation, for example) that can greatly affect the protein stability, pro-tumoral or anti-tumoral proteases such as metalloproteases (associated with cell migration), or caspases (involved in apoptosis) play an important role in protein degradation and are deregulated in cancer. The tumorigenesis does however also associate with alterations of systems involved in a more general control of proteins turn-over such as the Ubiquitin/Proteasome and the vesicular systems (caveolae, autophagosomes, lysosomes). A thousand E3 ubiquitin ligases have been described and some of them such as the Spk2, VHL, BRCA1, have indeed been shown to be deregulated in cancers. Various vesicular systems such as caveolae are involved in the regulation of various extracellular signals through the internalization and recycling of receptors such as PDGFR (Platelet Derived Growth Factor Receptor), EGFR (Epidermal Growth Factor Receptor) and TGFR (Transforming Growth Factor Receptor), G proteins such as H-Ras, Rac or Src, and PKC (Protein Kinase C) and may also participate to the tumorigenesis. Caveolines 1 and 2 are indeed suspected to act as tumor suppressors. Another vesicular system, the autophagy, may also play an important role in tumorigenesis but the benefits of the therapeutic targeting of this pathway is controversal. Finally, lysosomes functional alterations associated with overexpressed cathepsines (B, L and D) have been involved in the invasion and metastasis process and correlate with a poor prognosis for some tumors. Lysosomes may also be involved in cell death and the use of therapeutic agents destabilizing lysosomal membranes has been successfully tested on colorectal cancers cell lines.
Conclusion
The expression of a protein is controlled at the three DNA, messenger RNA and protein levels, but this is the structure of each of these three actors that determines the molecular mechanisms implemented for the synthesis, maturation and stability of the intermediate mRNA and the protein itself. The finding of structural abnormalities or functional impairments of these molecular mechanisms is essential to understand the causes of the pro- or anti-tumor protein aberrant expression levels in cancer. Therapeutic trials targeting some of these mechanisms have already proved their effectiveness as anti-tumorigenic agents. For example, the use of DNA methyl transferase (DNMT) inhibitors such as the 5-Aza-cytidine or of the proteasome inhibitor Bortezomib have been evidenced as two efficient ways to induce the reexpression of tumor suppressor genes.
1 Introduction
Les cellules tumorales acquièrent séquentiellement des propriétés (auto-suffisance en signaux de croissance, insensibilité aux signaux inhibiteurs de la croissance, échappement à la mort programmée par apoptose, potentiel replicatif illimité, angiogenèse, invasion tissulaire et métastase) qui leur procurent un avantage de croissance par rapport aux cellules normales dont elles dérivent. L'acquisition de ces propriétés s'accompagne d'une dérégulation d'expression et/ou de fonction de produits d'expression génique que l'on peut, de manière très simplifiée, séparer en deux grands groupes : (i) des facteurs pro-tumoraux (oncogènes, facteurs de croissance, récepteurs, régulateurs positifs du cycle cellulaire ...) dont l'activité et/ou l'expression est augmentée lors du processus tumoral ; et (ii) des facteurs anti-tumoraux (suppresseurs de tumeurs, régulateurs négatifs du cycle cellulaire, complexes contrôlant l'intégrité du génome, facteurs pro-apoptotiques, ...) qui à l'inverse sont le plus souvent sous-exprimés ou inactivés dans les tumeurs. La dérégulation de l'expression de ces gènes peut intervenir aussi bien au niveau du gène que de ses produits, l'ARN messager puis la protéine. L'ADN, support de l'information génétique, peut en effet être la cible de modifications épigénétiques, de mutations ponctuelles ou encore de remaniements chromosomiques. L'ARNm, intermédiaire entre gène et protéine, est également un maillon essentiel dans la dérégulation d'expression des gènes au cours de la tumorigenèse ce qui explique l'engouement qu'a suscité l'établissement de profils d'expression différentielle par puces nucléotidiques dans la recherche de marqueurs diagnostiques et pronostiques ou de cibles thérapeutiques [1]. Enfin, des altérations du contrôle de la traduction et/ou de la stabilité des protéines peuvent de la même manière être responsables de variations d'accumulation des protéines dans la cellule tumorale.
2 L'ADN
L'ADN peut être la cible de deux grands types d'altérations : (i) conformationnelles qui, suite à des modifications épigénétiques (méthylation, modifications post-traductionnelles des histones) perturbent notamment l'état de compaction de la chromatine et par conséquent l'accessibilité des facteurs de transcription et de la machinerie transcriptionnelle ; et (ii) nucléotidiques qui, sous l'influence d'une instabilité génétique peuvent conduire à des mutations ponctuelles jusqu'à d'importants remaniements chromosomiques.
2.1 Altérations conformationnelles
La méthylation de l'ADN est une modification covalente exercée par des ADN méthyltransférases (DNMT) sur les cytosines des di-nucléotides CpG concentrés dans de courtes régions appelées îlots CpG [2], et présents préférentiellement au niveau du promoteur et du premier exon de plus de 60% des gènes humains [3]. L'ADN est globalement sous-méthylé dans les cellules tumorales ce qui participerait à l'activation de gènes impliqués dans les processus d'invasion et de métastase comme la protéase PLAU (urokinase type plasminogen activator) dans les cancers du sein et de la prostate [4]. Cette hypo-méthylation peut également favoriser la prolifération cellulaire, notamment en affectant le gène codant l'IGF2 (Insulin-like growth factor) comme c'est le cas dans les tumeurs de Wilm's [5] (Fig. 1a) mais semble toutefois essentiellement participer à l'instabilité génomique régulièrement associée au processus cancéreux, en touchant les régions euchromatiques centromériques ou les séquences répétées de type LINE (long interspersed nuclear element) dans le cancer colorectal et les leucémies lymphoïdes chroniques par exemple [6] (Fig. 1b). Une hyper-méthylation peut cependant également être observée au niveau de loci particuliers (Fig. 1c). L'expression des méthylases DNMT1 et DNMT3B est d'ailleurs augmentée dans plusieurs types de cancers et est corrélée à l'hyper-méthylation des promoteurs de certains gènes suppresseurs de tumeurs comme les gènes codant p16INK4A, CDKN2A et 2B (cyclin-dependent kinase inhibitor), E-cadherin, MLH1 (human mismatch repair), RB1 (retinoblastoma) ou BRCA1 [7].
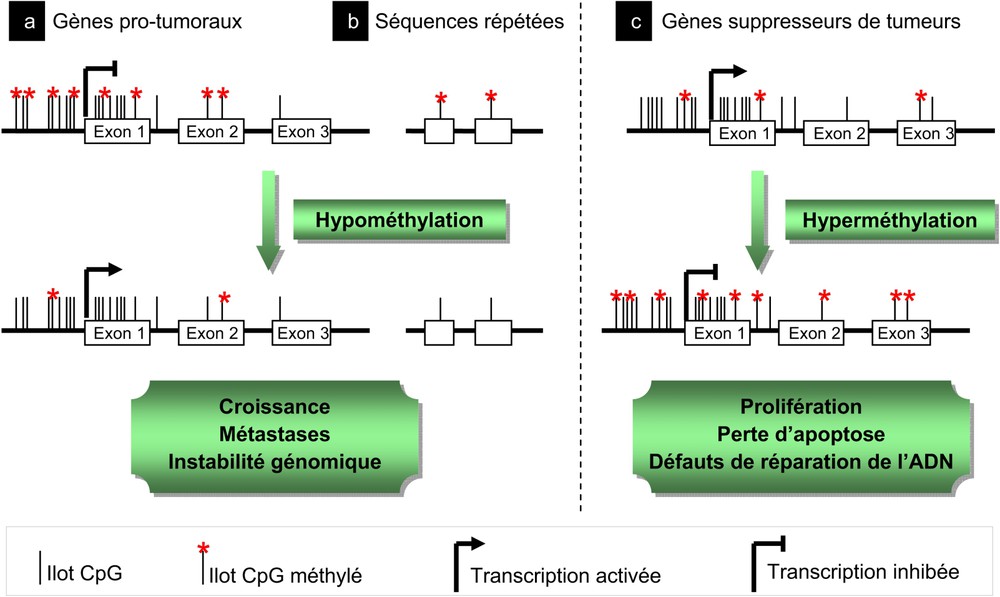
Différents types d'altération de la méthylation de l'ADN au cours de la tumorigenèse.
Des modifications post-traductionnelles touchant préférentiellement les résidus lysine des queues des histones H3 et H4 (désacétylation, méthylation, phosphorylation, SUMOylation, ...) et constituant le « code histone » modifient par ailleurs la structure de la chromatine en jouant sur l'interaction ADN–histones et/ou sur le recrutement de complexes transcriptionnels. Un rôle prépondérant des HDAC (histone deacetylase) dans la tumorigenèse est reflété d'une part par la levée de répression de gènes suppresseurs de tumeurs et d'autre part par la régression tumorale consécutives à l'inactivation de ces enzymes. Par exemple, HDAC1 est surexprimée dans les cancers de la prostate et associée à la répression du gène codant p21 par désacétylation des histones H3 et H4 présents sur son promoteur [8]. HDAC2 est quant à elle surexprimée dans les carcinomes gastriques et colorectaux et est également associée à une inhibition d'expression de p21 [9].
2.2 Altérations nucléotidiques
Le génome des cellules cancéreuses semble être particulièrement instable et la forte aneuploïdie retrouvée dans la plupart des cancers pourrait être l'élément déclencheur de la tumorigenèse avant l'accumulation successive de mutations [10]. On peut distinguer essentiellement trois grands types d'instabilité : NIN, MIN et CIN.
L'instabilité NIN ou nucléotidique, rare dans les cancers, est causée par des défauts dans les mécanismes de réparation de l'ADN de type NER (nucleotide excision repair) et BER (base excision repair) et concerne de subtiles changements dans la séquence ADN (substitutions, délétions, insertions de quelques nucléotides) regroupés sous le terme de mutations. Ceux-ci peuvent affecter la séquence codante du gène et conduire à une substitution d'acide aminé ou à l'expression d'une protéine tronquée. Les conséquences peuvent être multiples : altération fonctionnelle de la protéine (perte ou gain de fonction), modification de sensibilité à la dégradation et ainsi contribuer à la dérégulation de son niveau d'expression. Certaines mutations peuvent également affecter les séquences régulatrices des gènes. Elles aboutissent alors à une dérégulation du niveau de transcription suite à l'altération de la fixation de facteurs régulateurs. C'est, par exemple, le cas du polymorphisme (T en G) en position 309 dans le premier intron du gène HDM2, permettant le recrutement du facteur de transcription Sp1 et associé au risque de carcinome gastrique, liposarcome, cancer du sein et du cerveau [11]. A l'inverse, la mutation du site CDE/CHR (cell Cycle Dependent Element/cell Cycle Homology Region) un site présent dans le promoteur du facteur anti-apoptotique Survivine, empêche la liaison du répresseur et pourrait participer à sa surexpression observée dans de nombreux cancers [12]. Certaines mutations peuvent au contraire conduire à une réduction de l'activité promotrice. Le TGFbeta (Transforming Growth Factor) induit l'arrêt de croissance des cellules épithéliales notamment en induisant les inhibiteurs de cyclines p27KIP et p15INK4. Une mutation (A en G) en position –364 du promoteur du gène codant son récepteur de type 2 diminue son activité transcriptionnelle et pourrait participer à l'importante sous-expression, voire l'absence de ce récepteur dans plusieurs types de cancers [13].
L'instabilité MIN ou microsatellitaire, également rare dans les cancers, a lieu spécifiquement au niveau de régions répétées dans le génome comme les séquences polyA ou polyCA. Elle est causée par des défauts du système de réparation MMR (Mismatch Repair) et a notamment été associée au développement des formes héréditaires de cancer colorectal HNPCC (Hereditary Non-Polyposis Colorectal Cancer) [14]. Les loci responsables de cette instabilité résident au niveau des chromosomes 2 et 3, régions portant respectivement les gènes MSH2 et MLH1 [15,16].
L'instabilité CIN ou chromosomique semble plus universelle dans le processus cancéreux. Les bases moléculaires n'en sont pas clairement établies, mais les mécanismes impliqués dans la condensation des chromosomes, la stabilité des télomères, la formation et l'interaction chromosomes–microtubules ou encore le contrôle de la progression du cycle cellulaire (check-points) peuvent y participer. En particulier, des anomalies dans le point de contrôle du fuseau mitotique (spindle check-point), assurant la bonne répartition des chromosomes et la séparation des chromatides sœurs, ont été associées au phénomène de CIN, permettant aux cellules de sortir de mitose prématurément et de rentrer dans un nouveau cycle de duplication de leur ADN [17]. De plus, des altérations d'expression ou d'activité de certains acteurs de ce processus ont été retrouvées dans les cancers. Par exemple l'expression de MAD2 (Mitotic Arrest Deficient) est diminuée dans les cancers du sein [18] et BUB1 (Budding Uninhibited by Benzimidazoles) est muté dans les cancers colorectaux [19], ces deux protéines étant impliquées dans la formation du kinétochore, le point de jonction entre chromosome et fuseau mitotique. Un autre point de contrôle, celui des dommages à l'ADN (DNA-damage checkpoint) empêche l'entrée en mitose des cellules présentant des mutations et a également été impliqué dans cette instabilité. Les chromosomes porteurs d'ADN endommagé peuvent en effet ségréger anormalement ou être la cible d'altérations structurales causées par des cassures simple ou double brin, et plusieurs gènes impliqués dans ce point de contrôle ont été associés à la tumorigenèse. C'est le cas des gènes ATM (Ataxia Telangiectasia Mutated), BRCA1, BRCA2, et p53 [20,21]. Les remaniements qui en résultent sont très hétérogènes entre cancers y compris au sein d'un même type de cancer. Ils peuvent être classés en quatre grands groupes : (i) l'aneuploïdie, nombre anormal de chromosomes consécutif à une perte ou un gain de chromosomes entiers ou de bras chromosomiques qui reste l'une des propriétés les plus communes des cellules cancéreuses et corrélée à l'agressivité tumorale [22] (Tableau 1) : (ii) l'amplification de gènes qui a lieu préférentiellement au niveau de régions contenant des gènes dont la surexpression est associée à des stades plus ou moins précoces de la tumorigenèse et impliqués dans la croissance (ERBB2, EGFR, ...) la prolifération (c-Myc, N-Myc, K-Ras, Cycline D1, ...), l'inhibition de l'apoptose (Bcl2, HDM2, ...), la résistance aux drogues (MDR1, MPR ...) [23] ; (iii) la perte de gènes ou LOH (loss of heterozygosity) qui affecte différents gènes suppresseurs de tumeurs. C'est par exemple le cas des gènes codant p16 (CDKN2A) p15 (CDKN2B) et p53 ou encore de BRCA1 (Breast Cancer 1) et de son homologue BRCA2 [24] ; (iv) les réarrangements géniques qui sont dus à la translocation et la fusion de fragments de chromosomes. Ils conduisent souvent à l'expression de protéines chimériques et/ou placent le gène sous le contrôle du promoteur d'un autre gène entraînant, dans tous les cas, un niveau d'expression anormal. L'un des cas les plus étudiés est celui du chromosome de Philadelphie dû à la translocation de la partie 3′ de l'oncogène ABL (Abelson) localisé sur le chromosome 9 et sa fusion à la partie 5′ du gène BCR (Breakpoint Cluster Region) donnant naissance à la protéine de fusion BCR-ABL dans les leucémies myéloïdes chroniques [25]. La translocation t8,14 de l'oncogène c-Myc ne donne quant à elle pas naissance à une protéine chimérique mais place le gène sous la dépendance du promoteur de la chaîne lourde de l'immunoglobuline H (IgH) et est associée au développement de certaines leucémies et lymphomes, en particulier, du lymphome de Burkitt [26].
Analyse du niveau d'aneuploïdie dans divers types de cancers humains (d'après les données de Waever and Cleavland, 2006) [22].
| Tumeurs aploïdes | Tumeurs aneuploïdes | Tumeurs aneuploïdes | |
| Tumeurs solides | 9% | 72% | 19% |
| Sein | 15,5% | 70% | 14,5% |
| Colon | 0,7% | 86% | 13,3% |
| Poumon | 18% | 59% | 23% |
| Mélanome | 15% | 69% | 16% |
| Ovaire | 2,5% | 79% | 18,5% |
| Prostate | 8% | 70% | 22% |
| Astrocytome | 3,3% | 76% | 20,7% |
| Utérus | 4,7% | 60% | 35,3% |
| Tumeurs hématopoïétiques | 15% | 71% | 14% |
| Leucémie myéloïde aigue | 29% | 69% | 2% |
| Leucémie myéloïde chronique | 45% | 55% | 0% |
| Lymphome T-Cell | 8,5% | 88,5% | 3% |
| Lymphome de Burkitt | 53% | 46% | 1% |
| Lymphome folliculaire | 18% | 76% | 6% |
| Myélome multiple | 21% | 72% | 7% |
| Maladie d'Hodgkins | 11% | 55% | 34% |
3 L'ARN messager (ARNm)
Synthèse, maturation et stabilité des ARNm peuvent toutes être altérées lors du processus tumoral.
3.1 La transcription
La synthèse des ARNm (ou transcription) est un mécanisme cellulaire finement régulé qui requiert une part importante de la machinerie cellulaire puisque plus de 5% de nos gènes codent pour des facteurs de transcription [27]. La transcription peut, comme nous l'avons vu précédemment, être dérégulée au niveau de l'ADN par des modifications épigénétiques ou des mutations de sites de liaison pour des facteurs de transcription. Par ailleurs, quasiment toutes les voies de signalisation altérées dans les cancers aboutissent finalement à la dérégulation de l'expression et/ou de l'activité de facteurs de transcription et/ou de leurs cibles directes ou indirectes [28] (Fig. 2). De nombreux oncogènes et suppresseurs de tumeurs sont par ailleurs eux-mêmes des facteurs de transcription ou régulent des facteurs de transcription, leur dérégulation étant à l'origine de l'altération d'expression d'un ensemble de cibles impliquées dans l'acquisition de caractéristiques tumorales par les cellules. C'est notamment le cas de Myc, pRb/E2F ou p53 [29,30].
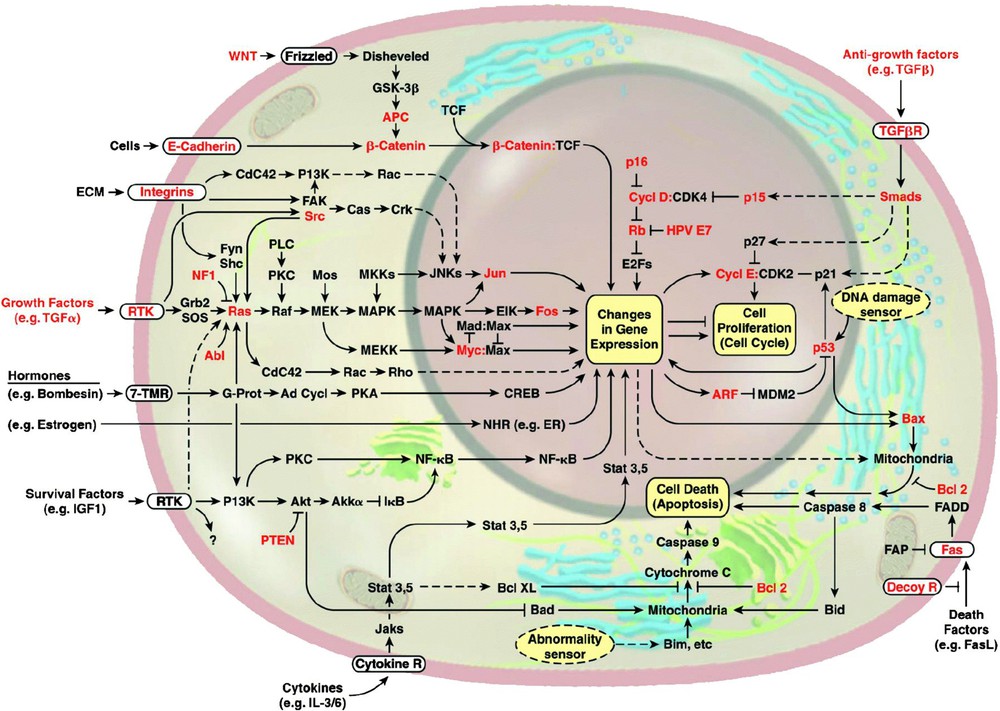
Voies de signalisation et principaux oncogènes ou suppresseurs de tumeurs (en rouge) impliqués dans la tumorigenèse (d'après Hanahan et Weinberg, 2000) [28]. Ces voies peuvent être dérégulées à plusieurs niveaux : ligand, récepteur membranaire, effecteurs. C'est par exemple le cas des voies de signalisation sous le contrôle de facteurs de croissance, avec notamment l'intervention de l'oncogène Ras ou encore de la voie Wnt/APC/β-caténine impliquée dans 80% des cancers colorectaux.
3.2 La maturation des ARNm
La maturation par épissage permet d'augmenter le nombre et la diversité des protéines. Plusieurs éléments cis-régulateurs sont impliqués dans ce processus : les sites d'épissage eux-mêmes, encadrant les exons (GT en 5′ et AG en 3′), et des séquences fortement dégénérées dites « enhancer » ou « silencer » reconnues par des facteurs trans-régulateurs respectivement de la famille SRP (serine (S) arginine (R) rich Protein) et hnRNP (heterogenous nuclear Ribo-Nucleo-Protein). Les cellules cancéreuses présentent régulièrement des défauts d'épissage conduisant à la sur-représentation d'un variant naturel, voire la création de variants cancer-spécifiques n'existant pas dans le tissu sain. Des mutations peuvent en particulier toucher les sites d'épissage comme la conversion d'AG en AT au niveau du site 3′ de l'exon 4 du gène APC conduisant à la perte de cet exon dans des tumeurs colorectales [31]. D'autres mutations peuvent à l'inverse créer de nouveaux sites d'épissage : (i) dans certains cancers du sein, la création d'un site 5′ aboutit à l'insertion de 69 nucléotides à l'ARNm du récepteur à l'oestrogène [32] ; et (ii) dans ce même type de tumeurs la création d'un site 3′ d'épissage ajoute 11 nucléotides à la séquence codante de l'ARNm de BRCA1, produisant une protéine tronquée [33]. Enfin, l'un des cas les plus étudiés a été celui de la protéine transmembranaire CD44, impliquée dans le contact cellule-cellule : plus de vingt formes différentes dues à l'inclusion de 10 exons alternatifs dans sa portion extracellulaire ont en effet été mises en évidence dans plusieurs types de cancer et associées notamment au potentiel métastatique [34]. Des dérégulations de facteurs trans-régulateurs tels que des protéines de la famille SRP (Serine Arginine Rich Protein) pourraient dans ce cas jouer un rôle dans l'inclusion des exons alternatifs de CD44 et par ce biais être associées au potentiel invasif des tumeurs. Ainsi, dans les tumeurs invasives du sein, l'augmentation d'expression de Tra2-B1 (Transformer-2-beta) est corrélée à l'inclusion des exons v4 et v5 de CD44 [35], et celle de SRP40 à l'inclusion des variants v2, v3, v5 et v6 [36]. Au contraire dans les adénocarcinomes du colon c'est la diminution de SRP40, SRP55 et SRP75 qui est corrélée à l'altération d'épissage de CD44 et au développement de métastases [37].
3.3 La stabilité de l'ARNm
La stabilité de la molécule d'ARNm est dépendante de sa région 3′UTR (UnTranslated Region). Cette région n'est pas parcourue par les ribosomes et constitue un lieu propice pour l'établissement d'interactions entre des séquences spécifiques de l'ARNm (riches en adénines et uridines nommées ARE (AU-Rich Element)) et des acteurs trans-régulateurs tels que des protéines, comme les ARE binding protein ou ARE-BP ou encore des ARN non codants comme les microRNA. Les ARNm contenant les ARE sont particulièrement labiles et aiguillés vers l'exosome, complexe multiprotéique à activité exonucléotidique responsable de leur dégradation [38]. Certains composants de ce complexe lient spécifiquement les séquences ARE et coopèrent avec d'autres protéines dirigeant les ARNm vers leur lieu de dégradation [39]. Plusieurs mécanismes peuvent être à l'origine d'une stabilisation de messagers codant des facteurs pro-tumoraux via ces séquences ARE : (i) des mutations. Des altérations des éléments ARE présents dans la région 3′UTR du messager de c-myc ont ainsi été décrites dans des lignées cellulaires issues de leucémies humaines et des cas de myélomes [40]. Néanmoins, la délétion de la portion 3′UTR du gène par recombinaison homologue dans un modèle murin ne conduit ni à la stabilisation de son ARNm, ni au développement de tumeurs remettant en question l'importance de cette région in vivo [41]. Des réarrangements ou troncations de la région 3′UTR de l'ARNm codant la cycline D1 ont par ailleurs été respectivement trouvés chez des patients atteints de lymphomes et dans des lignées cellulaires issues de cancer du sein [42] ; (ii) la dérégulation de protéines liant les éléments ARE (ARE-BP). L'une des plus étudiée est HuR qui, contrairement aux autres membres de sa famille (HuB, C, D) spécifiques du tissu neuronal, présente une expression ubiquitaire. Sa surexpression est observée dans plusieurs types de cancers et est notamment associée à une augmentation d'expression des oncogènes c-Fos et c-Myc, des cyclines A, B1 et D1, des facteurs de croissance EGF et VEGF, de la cyclo-oxygénase COX2 et de la métalloprotéase MMP9 [43]. HuD est quant à elle surexprimée dans certains cas de neuroblastomes et augmente la stabilité de l'ARNm codant l'oncogène N-Myc in vivo [44]. Son expression a également été observée dans un tissu tumoral non neuronal, le cancer du poumon à petites cellules [45]. Enfin, les microRNA (ou miRNA) sont d'autres éléments de régulation importants dans le contrôle de la stabilité des ARNm. Ils sont constitués en moyenne de 20 à 22 nucléotides et issus d'une digestion enzymatique de molécules d'ARN plus longues pré-miRNA par l'activité endonucléase de Dicer. Pris en charge par le complexe multiprotéique RISC (RNA induced silencing complex), ils s'associent par complémentarité préférentiellement à la région 3′UTR de leurs ARNm cibles et répriment ainsi leur traduction et/ou induisent leur dégradation [46]. Il semble que de nombreuses séquences codant pour des miRNA soient situées dans des régions chromosomiques associées au cancer, à proximité d'oncogènes, de suppresseurs de tumeur, ou de sites fragiles sensibles aux instabilités génétiques [47] ce qui pourrait expliquer les modifications de leur niveau d'expression dans les cellules cancéreuses. Par ailleurs, les niveaux d'expression de protéines impliquées dans leur synthèse (Dicer) et leur activité (Ago1, 3 et 4, constituants du complexe RISC) peuvent également être affectés dans les cancers [48]. Or, certains miRNA ont pour cibles des facteurs pro-tumoraux et, en contribuant à une diminution d'expression de ces derniers, se comportent eux-mêmes comme des suppresseurs de tumeurs. C'est le cas de Let-7 dont l'expression est diminuée dans les cancers du poumon non à petites cellules (NSCLC) et qui a pour cible l'ARNm de l'oncogène Ras [49]. Les microRNA miR-15a et miR-16-1 sont quant à eux délétés ou sous-exprimés chez 65% des patients atteints de leucémie lymphoïde chronique (CLL) et ont pour cible le facteur anti-apoptotique Bcl2 [50]. A l'inverse, certains miRNA sont surexprimés dans les cancers et jouent un rôle pro-tumoral. MiR-21, qui a en particulier pour cible le suppresseur de tumeur PTEN, est par exemple fortement exprimé dans divers types de cancers dont les glioblastomes, les cancers du sein et du pancréas [51].
4 La protéine
Les protéines constituent le produit final de l'expression des gènes et portent l'activité biologique. Leur concentration intracellulaire est dépendante des régulations précédentes mais également des mécanismes impliqués dans le contrôle de leur propre synthèse (traduction) et de leur stabilité qui peuvent également être la cible d'altérations dans les tumeurs.
4.1 La traduction
La traduction comporte trois étapes (l'initiation, l'élongation et la terminaison) chacune faisant intervenir ses propres facteurs de régulation : les facteurs eIF (eukaryotic Initiation Factor), eEF (eukaryotic Elongation Factor) et les facteurs de terminaison (release factor). L'étape d'initiation est l'étape limitante de la traduction. Elle dépend notamment de la disponibilité des facteurs eIF et des éléments de structure en 5′UTR et constitue une cible de dérégulation dans les cancers. Elle est elle-même divisée en deux phases : (i) la liaison de eIF4E à la coiffe 5′méthyl 7guanosine triphosphate (m7GTP) de l'ARNm permet la liaison de eIF4G et de eIF4A à activité hélicase, ces trois protéines formant le complexe eIF4F ; (ii) l'association de l'ARN de transfert porteur de la Méthionine (Met-ARNt) au facteur eIF2-GTP permet son transfert au niveau de la sous-unité 40S du ribosome en association avec les facteurs eIF3, eIF1A et eIF5 pour former le complexe de préinitiation 43S [52] (Fig. 3). eIF4E est surexprimé dans de nombreuses tumeurs (colon, sein, poumon, prostate, tractus gastro-intestinal, lymphomes, neuroblastomes) et est de mauvais pronostique pour les patients [53]. D'après Wendel et al. [54] eIF4E est d'ailleurs oncogénique in vivo. La surexpression des deux autres facteurs eIF4G et eIF4A impliqués dans la formation du complexe eIF4F a également été associée à la cancérogenèse de certains tissus comme le sein et le poumon [55,56], et à des cas de mélanomes et d'hépatocarcinomes [57,58] respectivement. eIF2 est un autre acteur clé de l'initiation. Responsable de l'apport du premier acide aminé méthionine de la chaîne peptidique, il forme un complexe ternaire avec l'ARNt-Met et le GTP. L'association à la sous-unité 40S du ribosome induit l'hydrolyse du GTP en GDP qui reste lié à eIF2. L'échange GDP en GTP sur eIF2 est assuré par eIF2B mais est inhibé lors de la phosphorylation de la sérine 51 de eIF2. Or cette phosphorylation est importante pour le maintien de l'intégrité de la cellule puisque l'expression artificielle d'une forme non phosphorylable de eIF2 inhibe l'apoptose et transforme les cellules NIH3T3 [59]. Le niveau d'expression de eIF2 et son taux de phosphorylation sont pourtant souvent élevés dans les cellules cancéreuses notamment dans les néoplasmes du colon [60]. Toutefois, même si l'initiation est dérégulée dans les cellules cancéreuses, on note rarement une augmentation globale de la traduction. En effet, la région 5′UTR de la majorité des ARNm est courte (moins de 200 bases), peu sujette aux structures secondaires et donc peu dépendante de l'activité hélicase du complexe eIF4F. Par contre, certains ARNm codant par exemple pour des facteurs oncogéniques, des facteurs de croissance ou des régulateurs de l'apoptose présentent une région 5′UTR plus longue et souvent riche en GC capable de former des structures secondaires [61]. Ces ARNm nécessitent une activité hélicase renforcée et sont donc plus dépendants de la formation du complexe eIF4F. C'est le cas de l'ARNm codant l'ornithine décarboxylase (ODC), FGF2 et VEGF deux régulateurs clés de la néoangiogenèse impliqués dans la progression tumorale et de la cycline D1 dont l'expression est fortement augmentée lors d'une surexpression d'eIF4E [62,63].
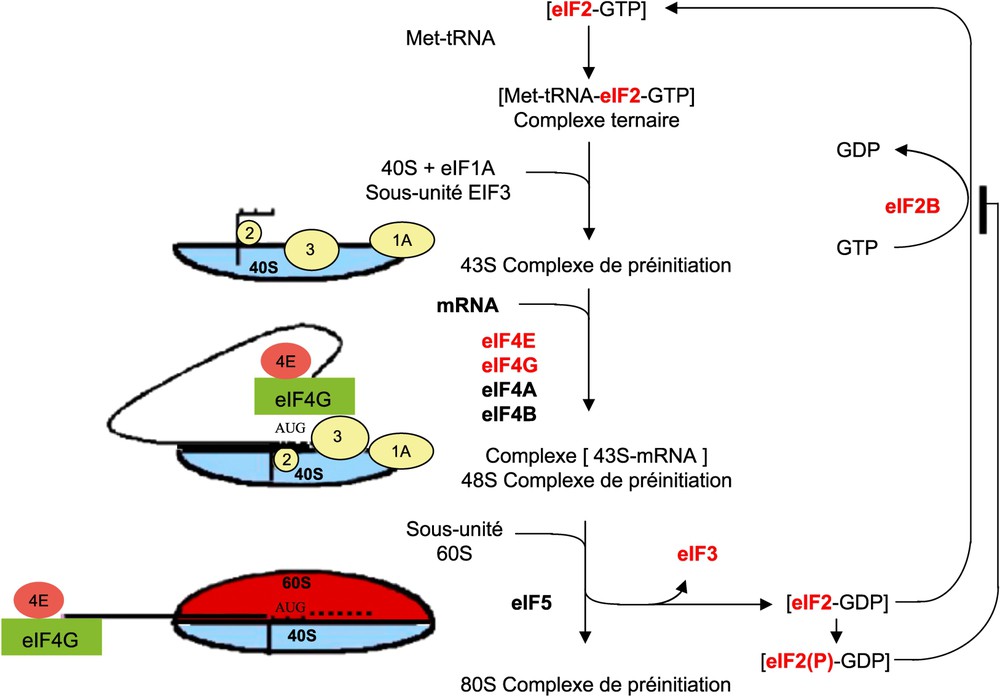
Mécanisme d'initiation de la traduction et facteurs limitants dérégulés dans les cancers (en rouge) (D'après Clemens, 2004) [52].
4.2 La stabilité et la dégradation des protéines
Le contrôle de la stabilité et de la dégradation des protéines constitue le dernier niveau de régulation permettant d'assurer des taux protéiques normaux. Les modifications post-traductionnelles comme la phosphorylation sont souvent importantes dans le contrôle de la stabilité des protéines. Des protéases pro-tumorales, comme des métalloprotéases (associées à la migration cellulaire), ou anti-tumorales telles les caspases (impliquées dans l'apoptose) [64] jouent par ailleurs un rôle important dans la dégradation protéique et sont dérégulées dans les cancers. Toutefois, la tumorigenèse peut également s'accompagner d'altérations affectant les systèmes contrôlant de manière plus générale le « turn-over » protéique. C'est le cas du système Ubiquitine/Protéasome et des systèmes vésiculaires impliqués dans le recyclage des protéines (caveolae, autophagosomes, lysosomes). Dans tous les tissus une grande partie des protéines intracellulaires est dégradée par le système Ubiquitine/Protéasome. Celle-ci se fait en deux étapes : une poly-ubiquitination des protéines cibles puis une reconnaissance et une dégradation par le protéasome. Trois protéines (E1, E2, E3) interviennent séquentiellement pour ajouter une chaîne d'Ubiquitines, polypeptide de 76 acides aminés, sur certains résidus Lysines de la protéine à dégrader : E1 sert d'activateur, E2 transporte les molécules d'Ubiquitine, et E3 porte l'activité enzymatique et assure la spécificité de substrat. Un millier d'enzymes E3 ont été décrites et diverses dérégulations affectant ces protéines ont été observées dans certains cancers. Par exemple, la surexpression de l'E3 ligase Spk2 spécifique du suppresseur de tumeur p27KIP s'avère oncogénique in vivo [65]. L'enzyme E3 VHL (Von Hippel–Lindau) est quant à elle mutée et inactivée dans certains cancers, notamment le rein, ce qui induit l'accumulation du facteur de transcription HIF-1 (Hypoxia Inducible Factor) et l'augmentation de la vascularisation des tumeurs [66]. Des mutations abolissant l'activité E3 ligase de BRCA1 ont également été retrouvées dans les cancers du sein familiaux et de l'ovaire [67]. Alors que l'accumulation de certaines cibles du système Ubiquitine/Protéasome résultant d'altérations fonctionnelles de protéines E3 semble pro-tumorale, l'inhibition du protéasome a pourtant été proposée dans le traitement des cancers. Les peptides boroniques acides dont fait notamment partie le Bortezomib, sont hautement sélectifs pour le protéasome et présentent une activité anti-tumorale dans des cancers hématologiques et solides en augmentant la sensibilité des tumeurs à la chimiothérapie ou en induisant l'arrêt de la croissance cellulaire [68]. Cette inhibition du protéasome s'accompagne d'une accumulation de p21 et de p27 [69]. Divers systèmes vésiculaires participent également, par fusion avec le compartiment endosomal/lysosomal, au recyclage et/ou à la dégradation des protéines et peuvent, comme le système ubiquitine-protéasome, être dérégulés dans des contextes de pathologie tumorale. C'est en particulier le cas des caveolae qui interviennent notamment dans la régulation de divers signaux extracellulaires en participant à l'internalisation et au recyclage de protéines associées ou contenues dans la membrane plasmique comme les récepteurs PDGFR (Platelet Derived Growth Factor Receptor), EGFR (Epidermal Growth Factor Receptor) et TGFR (Transforming Growth Factor Receptor), des protéines G telles que H-Ras, Rac ou Src, et les PKC (Protein Kinase C) [70]. Engelman et al. [71] proposent que les cavéolines 1 et 2, pourraient agir comme des suppresseurs de tumeur. Les gènes codant ces protéines sont en effet localisés au niveau de la région 7q31, une région souvent délétée dans les cancers. Une hyperméthylation des promoteurs de ces gènes a par ailleurs été décrite dans certaines lignées issues de cancer de la prostate [72]. Un autre système vésiculaire, l'autophagie, permettrait l'élimination des mitochondries lésées de façon à éviter le relargage néfaste de radicaux libres mais serait également déclenchée par la privation de nutriments et protègerait de la mort cellulaire en apportant à la cellule une source de constituants des macromolécules (acides aminés, acides gras, nucléotides). D'autres observations sont pourtant plutôt en faveur d'un rôle anti-tumoral puisque, poussée à l'extrême, l'autophagie conduit à la mort cellulaire par « autodigestion ». De plus, l'autophagie est stimulée par le suppresseur de tumeur PTEN [73] alors qu'elle est inhibée par les oncogènes mTOR et PKB/Akt [74]. Enfin, la Beclin 1, protéine participant à l'induction de l'autophagie, présente une activité anti-tumorale et le gène codant la Beclin1 est soumis à une LOH dans 40 à 75% des cancers du sein, de la prostate et de l'ovaire [75]. L'effet bénéfique du ciblage de cette voie dans la thérapie anticancéreuse fait néanmoins toujours débat. Des altérations fonctionnelles des lysosomes impliquant notamment certaines cathepsines ont également été associées aux mécanismes d'invasion et de métastase. La forte expression des cathepsines B, L et D est en particulier associée au mauvais pronostique de certaines tumeurs [76]. De plus, la localisation des lysosomes peut être altérée dans les cellules cancéreuses et passer de l'espace péri-nucléaire à proximité de la membrane plasmique avec laquelle ils fusionnent et libèrent leur contenu dans l'espace extracellulaire [77]. Les cathepsines agiraient alors de concert avec les métalloprotéases et le plasminogène pour dégrader la matrice extracellulaire et favoriser le pouvoir invasif des cellules tumorales. Enfin les lysosomes peuvent également être impliqués dans la mort cellulaire et l'utilisation thérapeutique d'agents déstabilisant les membranes lysosomales a d'ailleurs été testée avec succès sur des lignées cellulaires issues de cancers colorectaux [78].
5 Conclusion
La régulation de l'expression d'une protéine s'effectue aux trois niveaux que sont l'ADN, l'ARNm et la protéine, mais c'est la structure de chacun de ces trois acteurs qui conditionne les mécanismes moléculaires mis en jeu pour le contrôle de la synthèse, de la maturation et de la stabilité de l'ARNm intermédiaire ainsi que de la protéine elle-même. Dans ce contexte, la recherche d'anomalies de structure ou d'altérations fonctionnelles de ces mécanismes moléculaires est essentielle pour comprendre les causes des modifications du niveau d'expression de protéines pro- ou anti-tumorales dans les cancers. Des moyens d'intervention thérapeutiques ciblant certains de ces mécanismes ont en effet déjà prouvé leur efficacité anti-tumorale. Pour preuve, l'utilisation des inhibiteurs des DNA méthyl transférase (DNMT) comme la comme la 5-Aza-cytidine ou encore l'utilisation d'inhibiteurs du protéasome tel que le Bortezomib, deux moyens d'induire la réexpression de gènes suppresseurs de tumeurs.
Remerciements
Les auteurs tiennent à remercier la Ligue contre le Cancer et l'Association pour la Recherche contre le Cancer (ARC) pour leur soutien financier ainsi que les Dr Hanahan, Weinberg et Clemens pour leur autorisation concernant l'utilisation des Figs. 2 et 3 précédemment publiées dans les revues Cell et Oncogene.