



 CC-BY 4.0
CC-BY 4.0
1. Introduction
La maladie de Parkinson (MP) est la deuxième maladie neurodégénérative la plus fréquente après la maladie d’Alzheimer. Le nombre d’individus atteints dans le monde a doublé de 1990 à 2016 pour atteindre 6 millions d’individus aujourd’hui [1], et il est anticipé que 12 millions d’individus seront atteints en 2040, du fait du vieillissement de la population [2]. Le coût économique de la maladie est estimé à 23 milliards de dollars par an aux États-Unis (21 milliards d’euros), et les projections tablent sur un doublement à 50 milliards en 2040 (46 milliards d’euros) [3]. Sur le plan clinique, la MP se caractérise par des symptômes moteurs (bradykinésie, rigidité extra-pyramidale et tremblement de repos) ainsi que par des symptômes non moteurs (constipation, hyposmie, dépression, troubles cognitifs et du sommeil principalement) d’évolution progressive. Les symptômes moteurs débutent typiquement après 60 ans et l’espérance de vie au diagnostic est d’environ 15–20 ans [4]. Elle touche plus les hommes que les femmes, avec un ratio de 1,5. La maladie est caractérisée neuropathologiquement par une perte progressive qui touche préférentiellement les neurones dopaminergiques de la substance noire, et qui s’accompagne de corps de Lewy dont un composant majeur est l’alpha-synucléine.
Dans de nombreux cas l’étiologie de la MP est multifactorielle associant facteurs génétiques et environnementaux, il s’agit de la maladie de Parkinson idiopathique. Une des plus récentes études d’association pangénomique a identifié plus de 90 locus génétiques de susceptibilité à la maladie [5]), dont chacun a un effet très faible. Dans d’autres cas, il existe une cause majeure à la maladie : intoxication aux pesticides comme rare cause environnementale ou forme monogénique dans laquelle un variant pathogène dans un seul gène suffit à causer la maladie.
À ce jour, une douzaine de gènes sont responsables de formes monogéniques de MP [6]. L’existence de formes familiales dans lesquelles des mutations dans ces gènes sont exclues indique que d’autres gènes restent encore inconnus.
Les caractéristiques cliniques et physiopathologiques des patients avec une cause génétique diffèrent généralement de ceux avec une cause multifactorielle. Par exemple, les patients avec un variant pathogène du gène Leucine-rich repeat kinase 2 (LRRK2) ont une progression plus lente, une survie plus longue et moins de symptômes moteurs que les MP classiques [7], tandis que les patients porteurs d’un variant pathogène du gène Glucosidase, beta, acid 1 (GBA1) ont une évolution plus rapide avec des troubles cognitifs plus précoces [8]. Les patients avec des variants pathogènes bi-alléliques du gène Parkin (PRKN) ont un début précoce (en moyenne autour de 25–35 ans), une évolution très lente et une très bonne réponse à la L-dopa [9]. La fréquence de mutations dans ces gènes parmi les patients atteints de MP varie. Elle est faible pour Synuclein alpha (SNCA) et PRKN (de 1 pour 1000 à 1 pour 100 patients parkinsoniens), modérée pour LRRK2 et élevée pour GBA1 dans les populations d’origine européenne (environ 2,5 % et de 7 à 12 %, respectivement) [10, 11, 12] et élevée pour LRRK2 chez les Juifs Ashkénazes et Nords Africains (20 à 40 %) [13, 14]. En population générale d’origine européenne, la fréquence cumulée des porteurs de mutations de variants à risque de GBA1 est d’environ 4 %, contre 4/10 000 pour le variant de loin le plus fréquent de LRRK2 (G2019S). Elle est extrêmement faible pour SNCA et PRKN. Il est important de noter ici que la pénétrance (i.e. le risque de développer la maladie au cours de la vie chez un individu porteur) varie selon le gène. Elle est complète pour SNCA et PRKN, mais seulement de l’ordre de 10 à 30 % pour LRRK2 et GBA1 [15, 16, 17]. Ainsi, même si la fréquence des variants dans ces deux derniers gènes est relativement élevée en population générale, seule une minorité de ces individus développeront une maladie de Parkinson. Il est donc plus précis de considérer les individus porteurs de variants dans ces gènes comme étant à « haut risque » de développer la maladie de Parkinson, ce qui pose des difficultés en termes thérapeutiques comme nous le verrons plus loin.
À ce jour, il n’existe aucun traitement disponible qui permette de ralentir l’évolution naturelle de la maladie, encore moins de traitement curatif. La stratégie thérapeutique principale vise à compenser la perte des neurones dopaminergiques par l’administration du précurseur de la dopamine, la L-dopa, ou encore d’agonistes dopaminergiques. L’intensité des symptômes augmentant progressivement, les posologies doivent être augmentées avec des effets indésirables fréquents. Il est donc urgent de trouver des traitements capables de ralentir, voire stopper, l’évolution naturelle de la MP. Les mécanismes physiopathologiques qui sous-tendent le rôle de certains gènes responsables de formes monogéniques constituent des cibles thérapeutiques intéressantes. Fondées sur la compréhension des mécanismes en jeu dans les formes génétiques, de nouvelles approches thérapeutiques sont déjà à l’essai qui pourraient ouvrir la voie à la médecine de précision dans la MP.
Les gènes connus de MP n’ont pas tous la même fonction et leurs mutations les mêmes conséquences, ce qui explique des phénotypes différents. Par exemple, les variants pathogènes du gène PRKN affectent principalement la mitochondrie, notamment les voies de sa dégradation (mitophagie), tandis que les variants pathogènes du gène GBA1 altèrent essentiellement le fonctionnement du lysosome (cf. infra). Il s’agit donc d’aller vers une médecine de précision fondée sur le mécanisme de la pathologie. De plus, elle devra être personnalisée, car l’idée sera de la délivrer aussi au meilleur moment au cours de la maladie, voire avant l’apparition des symptômes. Enfin, les différents mécanismes physiopathologiques en lien avec un gène spécifique peuvent aussi jouer un rôle dans la MP idiopathique (patients sans cause génétique) avec l’espoir qu’à terme, une thérapie ciblée efficace pour une forme de MP génétique particulière puisse bénéficier plus largement aux autres patients.
Nous nous proposons de centrer cette revue sur les quatre gènes majeurs de MP qui sont la cible de thérapeutiques en cours d’évaluation : SNCA, GBA1, LRRK2 et PRKN.
2. SNCA et l’alpha-synucléine
SNCA a été le premier gène de MP monogénique découvert dès 1997 [18]. Il a permis de comprendre le rôle central joué par l’alpha-synucléine dans la physiopathologie de la maladie [19]. En effet l’alpha-synucléine est la protéine majoritaire des corps de Lewy, principaux marqueurs anatomo-pathologiques de la maladie.
Les variants faux-sens pathogènes de SNCA prédisposent à l’accumulation et à l’agrégation d’alpha-synucléine (Figure 1). Il existe également une propagation de cellule en cellule de l’alpha-synucléine pathologique dans la MP [20] qui pourrait rendre compte de l’extension spatiale des lésions. Les duplications et les triplications de SNCA augmentent l’expression de l’allèle sauvage avec un effet de dosage génique [21]. Ces observations démontrent que la simple surexpression de la protéine normale est suffisante pour produire la maladie de Parkinson. Si la toxicité de l’alpha-synucléine est établie, il n’est pas encore démontré sous quelle forme (monomère, oligomère ou fibrille) celle-ci se manifeste. De plus, il est important de préciser que les corps de Lewy dont le composant majoritaire est l’alpha-synucléine sont présents chez la majorité des patients avec une MP idiopathique. Ainsi, les thérapeutiques présentées ci-dessous ne concernent pas uniquement les MP liées à des variants pathogènes de SNCA mais pourraient également s’appliquer aux MP idiopathique.
Représentation de la physiopathologie associée à l’alpha-synucléine (SNCA) et des stratégies thérapeutiques à l’essai.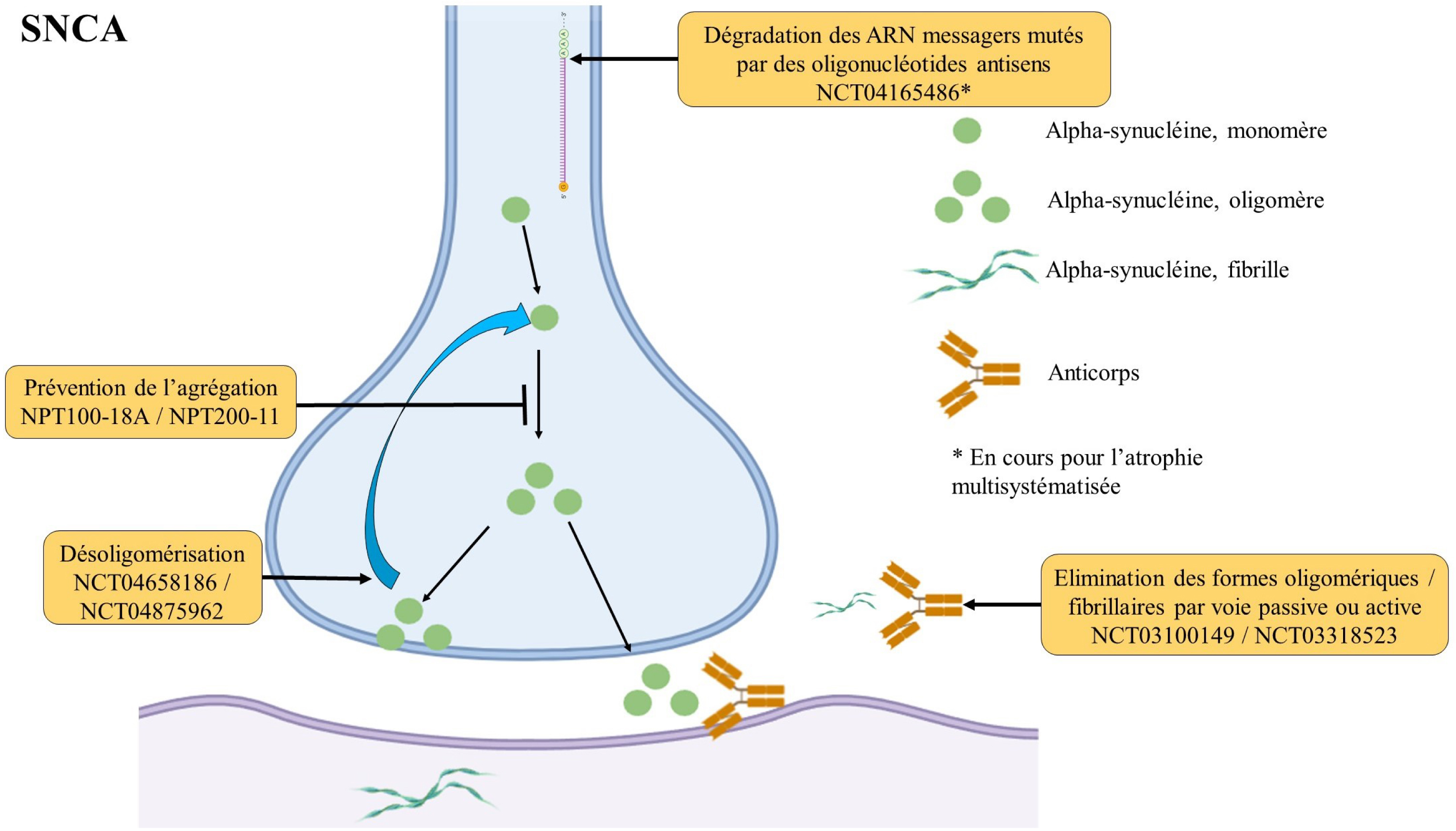
Une première stratégie consiste à utiliser des thérapeutiques capables de prévenir l’agrégation des molécules d’alpha-synucléine mal conformées sans altérer les fonctions normales de l’alpha-synucléine en amont. Les inhibiteurs de la conformation anormale de l’alpha-synucléine NPT100-18A et NPT200-11 ont montré une efficacité dans des modèles transgéniques murins de MP surexprimant l’alpha-synucléine [22] (Figure 1). Le Minzasolmin (UBC0599), un dérivé de ces molécules, est capable de ramener à une forme monomérique les alpha-synucléines oligomériques liées à la membrane en les détachant de celle-ci [23]. Cette molécule est actuellement en essai thérapeutique de phase 2 (NCT04658186) après un essai de phase 1 encourageant (NCT04875962) (Tableau 1).
Essais thérapeutiques ciblés passés et présents
| Substance active | Sponsor | Gène cible | Mécanisme d’action | Identifiant de l’essai clinique | Phase | Objectif primaire | Début | Résultats | Fin de l’essai |
|---|---|---|---|---|---|---|---|---|---|
| PD03A | Affiris AG | Immunisation active | NCT02267434 | 1 | Sécurité et tolérance | 2014 | Bonne tolérance, réponse immune dose dépendante | 2017 | |
| BIIB054 | Biogen | Immunisation passive | NCT02459886 | 1 | Sécurité et tolérance | 2015 | Bonne tolérance | 2017 | |
| BIIB054 | Biogen | SNCA | Immunisation passive | NCT03318523 | 2 | Evolution du score total MDS-UPDRS à 72 semaines | 2018 | Pas d’efficacité clinique comparativement au groupe contrôle | 2021 |
| Prasinezumab/PRX002 | Hoffmann-La Roche | Immunisation passive | NCT03100149 | 2 | Evolution du score total MDS-UPDRS à 52 semaines | 2017 | ND | 2026 | |
| Minzasolmin/UCB0599 | UCB Biopharma SRL | Désoligomérisation | NCT04658186 | 2 | Sécurité, tolérance, évolution du score totale MDS-UPDRS à 18 mois | 2020 | ND | 11/2024 | |
| Venglustat | Genzyme | Molécule chaperone | NCT02906020 | 2 | Sécurité, tolérance, évolution du score total MDS-UPDRS à 52 semaines | 2016 | Détérioration de la fonction motrice dans le groupe traité | 2022 | |
| Ambroxol | Lawson Health Research Institute | Molécule chaperone | NCT02914366 | 2 | Evolution des scores ADAS-cog et CGIC à 26 et 52 semaines | 2015 | ND | 2025 | |
| Ambroxol | University College London | GBA1 | Molécule chaperone | NCT02941822 | 2 | Niveau de Gcase dans le LCR à 186 jours comparé à la baseline | 2016 | Ambroxol détecté dans le LCR, augmentation de l’activité de la Gcase | 2018 |
| Ambroxol | Fondazione I.R.C.C.S. Istituto Neurologico Carlo Besta | Molécule chaperone | NCT05287503 | 2 | Evolution du score MOCA et des troubles cognitifs à 52 semaines | 2022 | ND | 2024 | |
| Ambroxol | University College London | Molécule chaperone | NCT05778617 | 3 | Evolution du score total MDS-UPDRS à104 semaines | 2023 | ND | 2028 | |
| LY3884961 | Prevail Therapeutics | Thérapie génique | NCT04127578 | 1/2a | Sécurité, tolérance, immunogénicité | 2020 | ND | 2029 | |
| BIIB094 | Biogen | Oligonucléotide antisens | NCT03976349 | 1 | Sécurité et tolérance | 2019 | ND | 08/2024 | |
| DNL151/BIIB122 | Biogen | Inhibition de la Kinase LRRK2 | NCT04056689 | 1 | Sécurité et tolérance, pharmacocinétique et pharmacodynamique | 2020 | Bonne tolérance et meilleure pharmacocinétique que DNL201 | 2020 | |
| DNL151/BIIB122 | Biogen | LRRK2 | Inhibition de la Kinase LRRK2 | NCT05348785 | 2 | Sécurité et évolution du score MDS-UPDRS parties II et III à 144 semaines | 2022 | ND | 2025 |
| DNL201 | Denali Therapeutics Inc. | Inhibition de la Kinase LRRK2 | NCT03710707 | 1 | Sécurité et tolérance, pharmacocinétique et pharmacodynamique | 2018 | Bonne tolérance et sécurité, inhibition de LRRK2 | 2019 | |
| DNL201 | Denali Therapeutics Inc. | Inhibition de la Kinase LRRK2 | NCT04551534 | 1 | Sécurité et tolérance, pharmacocinétique et pharmacodynamique | 2017 | Bonne tolérance et sécurité, inhibition de LRRK2 | 2018 |
MDS UPDRS Movement Disoder Society — Unified Parkinson’s Disease Rating Scale; ADAS-cog Alzheimer’s Disease Assessment Scale-Cognitive Subscale; CGIS Clinical Global Impression Scale; LCR: Liquide Céphalo-Rachidien; MOCA: Montréal Cognitive Assessment Scale; ND: Non Disponible.
Une seconde stratégie repose sur l’utilisation d’anticorps anti alpha-synucléine avec pour objectif de diminuer le taux de la protéine (Figure 1). Il s’agit d’anticorps monoclonaux ciblant différentes parties de la protéine. L’objectif est d’augmenter, via un processus d’immunité, l’élimination des formes oligomériques et/ou fibrillaires d’alpha-synucléine, et donc in fine de réduire sa toxicité cellulaire et sa diffusion pathologique [24, 25]. Deux approches coexistent : une immunisation active contre la forme agrégée par vaccination (PD01A et PD03A, [26]) et une immunisation passive par l’injection d’anticorps monoclonaux (BIIB054, BAN0805, PRX002, [27]). Les premiers résultats ont montré un engagement de la cible ainsi qu’une sécurité suffisante (NCT02267434, NCT02459886), ce qui a permis la réalisation de deux essais de phase 2 contrôlés et randomisés (NCT03318523 et NCT03100149) (Tableau 1). Néanmoins, ces approches par anticorps visant essentiellement les agrégats d’alpha-synucléine extracellulaire sont probablement inefficaces dans le ciblage de l’alpha-synucléine intracellulaire [28]. Une autre approche permettant de surmonter cet inconvénient consiste à utiliser des oligonucléotides antisens. Les oligonucléotides antisens ont la propriété d’induire la dégradation de l’ARN messager cible intracellulaire et, ainsi, de réduire le niveau de la protéine correspondante. Leur efficacité dans l’initiation de l’agrégation et dans la progression spatiale des inclusions d’alpha-synucléine a été démontrée in vivo dans des modèles animaux de MP [29, 30, 31]. Aucun essai clinique utilisant des oligonucléotides antisens ciblant les ARN messagers de SNCA n’est en cours. Néanmoins, un essai clinique a débuté dans l’atrophie multisystématisée, une maladie proche de la MP sur le plan physiopathologique (NCT04165486) (Figure 1).
3. GBA1
Les variants bialléliques pathogènes du gène GBA1 sont à l’origine de la maladie de Gaucher [32], une maladie rare affectant le lysosome. Plusieurs auteurs ont observé que certains patients avec une maladie de Gaucher présentent un syndrome parkinsonien et que les parents de patients avec une maladie de Gaucher ont une MP plus fréquemment qu’attendu [33]. L’association entre variants responsables de maladie de Gaucher et MP a été démontrée dans une étude internationale [34]. De nombreux variants rares ont été observés dans le gène GBA1 dont certains, associés à la maladie de Gaucher, sont qualifiés de sévères alors que les autres ont un effet modéré. Sur le plan clinique, les patients MP-GBA1 ont une maladie plus sévère avec un début plus précoce, une plus grande fréquence de symptômes non moteurs et une évolution plus rapide des symptômes moteurs et non moteurs que dans la MP idiopathique [8]. De plus, ils ont un déclin cognitif plus important et une plus grande fréquence de troubles psychiatriques (anxiété, dépression et hallucination, etc. [35]). Ces différences sont d’autant plus marquées que les patients sont porteurs de mutations dites sévères. Par ailleurs, la stimulation cérébrale profonde, efficace chez les patients MP-PRKN, semble aggraver les troubles cognitifs chez les patients MP-GBA1 [36].
GBA1 code pour la Beta-glucocérébrosidase (Gcase), une enzyme située au niveau de la membrane lysosomale et responsable de la dégradation de plusieurs glycolipides, principalement le glucosylcéramide (Figure 2). La Gcase est synthétisée dans le réticulum endoplasmique et ensuite transportée jusqu’au lysosome où elle va exercer son activité enzymatique [37]. Globalement, deux mécanismes physiopathologiques sont décrits lorsqu’un variant pathogène hétérozygote est présent dans GBA1 : soit il entraîne une perte de la fonction enzymatique, soit il diminue l’adressage de la Gcase du réticulum endoplasmique jusqu’à la membrane du lysosome. Les deux mécanismes aboutissent à une accumulation toxique des substrats de la Gcase [38, 39], et le second mécanisme produirait également une altération du fonctionnement global du réticulum endoplasmique [40]. Compte tenu de ces différents mécanismes, il existe actuellement trois stratégies pour restaurer la fonction de GBA1 : les thérapies de remplacement enzymatique, les thérapies de réduction du substrat et enfin les thérapies géniques.
Représentation de la physiopathologie associée à la Beta-glucocérébrosidase (GBA1) et des stratégies thérapeutiques à l’essai.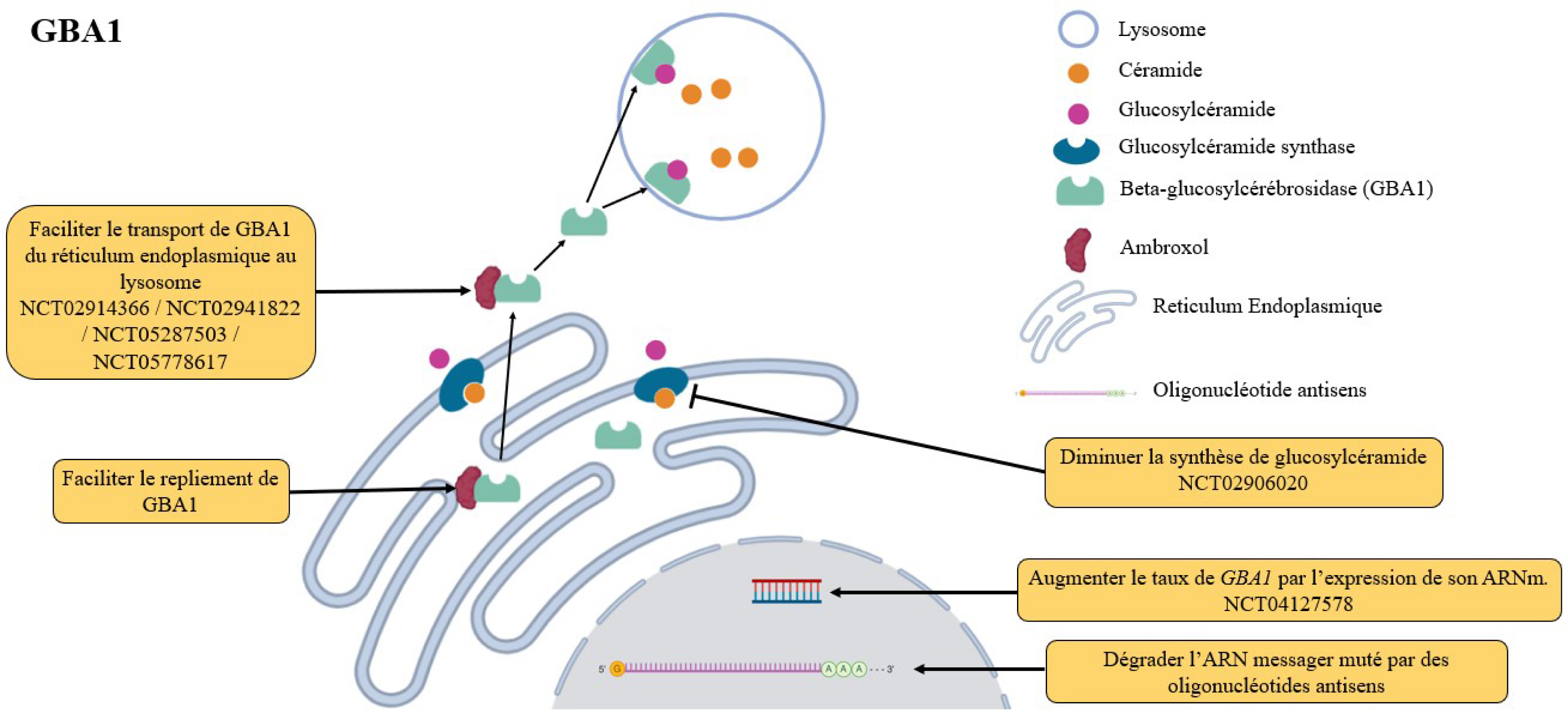
3.1. Les thérapies de remplacement enzymatique
Les thérapies de remplacement enzymatique visent à délivrer la forme active de l’enzyme non fonctionnelle. À ce jour, elles sont principalement utilisées dans la maladie de Gaucher non neuropathique (type 1), car l’enzyme administrée en périphérie ne pénètre pas dans le système nerveux [41]. Elles ne sont pas efficaces dans la MP-GBA1 en raison de l’absence de passage au travers de la barrière hémato-encéphalique [42], il faudrait donc envisager de délivrer l’enzyme directement dans le système nerveux pour atteindre la cible de la pathologie.
3.2. Les thérapies de réduction du substrat
3.2.1. Deux options sont à l’essai
La première option consiste à diminuer la quantité de glucosylcéramide (le principal substrat toxique) au moyen d’inhibiteurs de la glucosylcéramide synthase tels que le venglustat [38] (Figure 2). Après des essais précliniques et de phase 1 encourageants, un essai de phase 2 du venglustat a montré une diminution de la quantité de glucosylcéramide (NCT02906020) [43]. Malheureusement, les patients sous venglustat se sont détériorés plus rapidement sur le plan moteur, cognitif et psychiatrique que le groupe placebo, ce qui a interrompu le développement de cette molécule dans la MP (Tableau 1).
La deuxième option consiste à restaurer la fonction GCase en facilitant son adressage à la membrane lysosomale. Cette stratégie s’appuie sur des molécules chaperonnes qui ont la propriété de faciliter la conformation correcte d’enzymes anormalement repliées et leur transport. Parmi les molécules chaperonnes à l’essai pour la maladie de Gaucher, on peut citer l’isofagomine, le miglustat, l’éliglustat, le NN-DNJ et l’ambroxol [44]. Ces molécules ont la capacité de traverser la barrière hémato-encéphalique, de se lier à la Gcase dans le réticulum endoplasmique et de faciliter son repliement et son transport jusqu’au lysosome (Figure 2). Les études sur modèles animaux et cliniques ont montré que la plus efficace est l’ambroxol [45]. Elle augmente l’activité et le niveau de GCase, réduit la quantité de substrat, augmente la translocation au lysosome et agit comme un concurrent des inhibiteurs de GCase dans différents modèles animaux ainsi que chez l’humain. Une étude de phase 2 a montré la sécurité et la bonne tolérance de l’ambroxol chez les patients Parkinsoniens (NCT02941822) (Tableau 1). Deux autres essais de phase 2 sont en cours (NCT02914366, NCT05287503) et un essai de phase 3 est en cours d’initiation (NCT05778617) (Tableau 1).
3.3. Les thérapies géniques
Les thérapies géniques, c’est-à-dire l’administration directe du gène fonctionnel, sont à un stade plus expérimental pour GBA1. L’administration du gène GBA1 se fait par un vecteur viral, ce qui a montré une certaine efficacité chez les modèles murins via des administrations intracérébrales [46] et systémiques [47, 48] (Figure 2). Sur ce principe, un essai multicentrique de phase 1/2a est en cours avec administration intracisternale de différentes doses de la molécule nommée LY3884961 chez des MP-GBA1 (NCT04127578) (Tableau 1). Les premiers résultats sont attendus pour 2029.
Contrairement à LRRK2 (cf. plus bas), l’activité de GBA1 ne semble pas être diminuée chez les patients parkinsoniens sans variant GBA1 comparativement à des individus sains [49]. Les traitements ciblant l’activité GCase concerneront donc, probablement, uniquement les patients avec des variants GBA1, qui représentent néanmoins un contingent important de patients parkinsoniens (entre 7 % et 12 % selon les études réalisées en population européenne) [10, 11, 12].
4. LRRK2
Les variants pathogènes du gène LRRK2 sont la cause la plus fréquente de forme monogénique de la maladie de Parkinson. Le variant pathogène G2019S de LRRK2 rend compte de plus de 1 % des cas avec MP dans les populations européennes et de 20 à 40 % des cas chez les Juifs Ashkenazes et les Nord-Africains en raison d’un effet fondateur [13, 14]. Néanmoins, la pénétrance est incomplète et varie beaucoup selon les études et les populations, de 14 à plus de 80 % à 80 ans [7, 17]. Six autres variants pathogènes du gène LRRK2 sont décrits, tous localisés dans les domaines catalytiques kinase ou GTPase de LRRK2. Ces variants ont pour point commun d’entraîner une augmentation de l’activité kinase de LRRK2 et donc son autophosphorylation et celle de ses substrats, dont des protéines de la famille des Rab GTPases (par exemple Rab10 et Rab8). Cette augmentation est soit directe, soit indirecte via une augmentation de l’activité GTPase qui elle-même va augmenter l’activité Kinase [50, 51]. Les altérations de protéines Rab en lien avec les mutations de LRRK2 sont variées et dépendent du type de protéine Rab. Elles vont de la phosphorylation de sites aberrants (RAB8A, RAB7L1) à l’augmentation de la phosphorylation (RAB10, RAB35, RAB123, RAB129) en passant par leur dysfonctionnement médié par la diminution de l’activité protéique (RAB7) [52]. Les protéines Rabs contrôlent l’agrégation, la toxicité et la sécrétion d’alpha-synucléine (Rab8b, Rab11a, Rab13 et Rab39b [53]). Elles promeuvent l’élimination des inclusions d’alpha-synucléine et réduisent la toxicité induite par cette protéine. L’expression de Rab11a et Rab13 améliore aussi la sécrétion d’alpha-synucléine et son recyclage endocytique dans les cellules en réponse à une accumulation d’alpha-synucléine.
Des traitements visant à réduire l’activité kinase de LRRK2 de manière directe ou indirecte sont ainsi à l’essai (voir par exemple le tableau 2 dans [51]). Les essais précliniques d’inhibiteurs du domaine kinase (DNL151/BIIB122, DNL201, GNE-7915) indiquent qu’ils ont une bonne capacité à traverser la barrière hémato-encéphalique et une fonction neuroprotectrice in vitro sur des modèles cellulaires et in vivo chez le rongeur [54, 55]. Bien que les premières études cliniques de phase 1 des inhibiteurs de LRRK2 aient été limitées par une toxicité pulmonaire, la tolérance des essais suivants a été bonne (NCT04056689, NCT04551534, NCT03710707) (Figure 3, Tableau 1). Le DNL151/BIIB122 est actuellement en essai de phase 2 (NCT05348785). La difficulté rencontrée pour ces inhibiteurs de l’activité kinase ciblés sur LRRK2 reste leur spécificité, étant donné que les kinases ont en général un haut degré d’homologie de séquence et de structure.
Représentation de la physiopathologie associée à LRRK2 et des stratégies thérapeutiques à l’essai.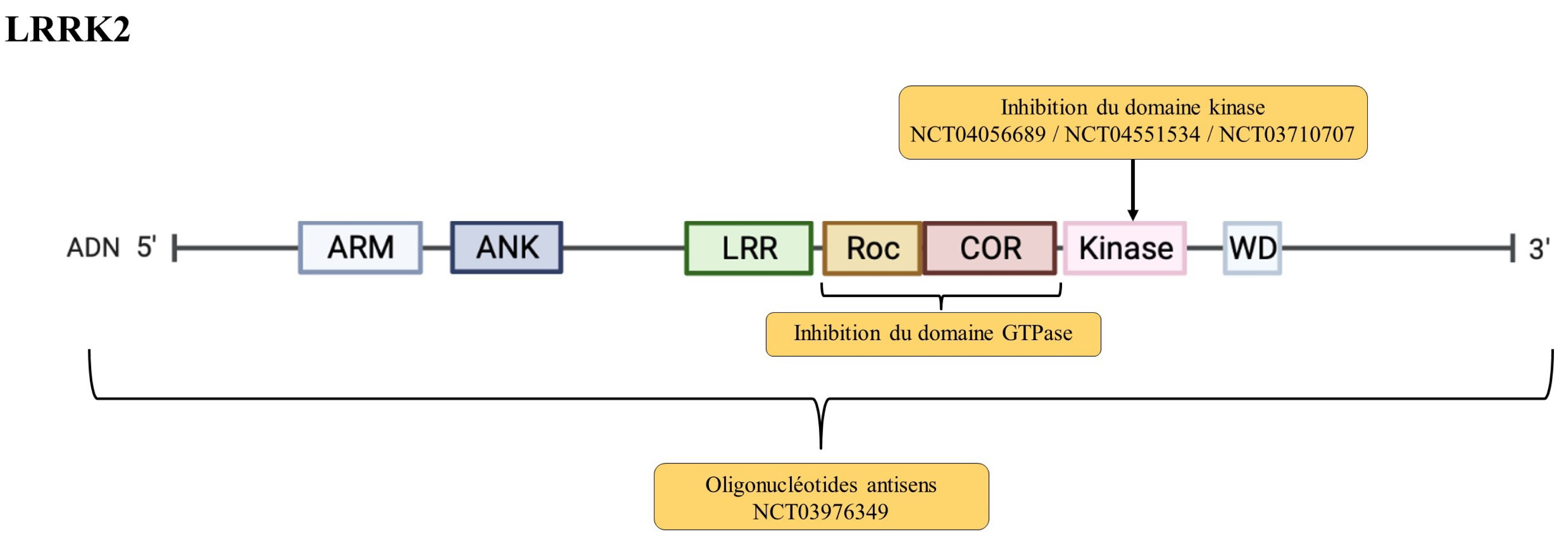
En plus du ciblage direct de l’activité kinase, les inhibiteurs de la liaison du GTP agissant sur la fonction GTPase (elle-même favorisant l’activité kinase) pourraient être une méthode efficace d’inhibition de LRRK2. Trois molécules inhibitrices font actuellement l’objet d’essais précliniques [56].
Au-delà du ciblage de la fonction de LRRK2, les récents progrès en médecine de précision et en thérapie génique offrent la possibilité d’inactiver directement la production de la protéine LRRK2 mutée et donc de réduire son activité kinase excessive. Différentes stratégies sont à l’œuvre : le clivage de l’ARN messager par un petit ARN en épingle à cheveux (hairpin RNA) [57], la dégradation de l’ARN messager par un oligonucléotide antisens [58], la modification de l’épissage pour exclure l’exon portant le site actif par un oligonucléotide antisens ciblant l’épissage de cet exon [59], ou encore la dégradation directe de la protéine synthétisée par des petites molécules chargées d’adresser la protéine dysfonctionnelle à la machinerie de dégradation protéique de la cellule via le recrutement d’une ubiquitine ligase E3 [60]. Les résultats précliniques, réalisés à la fois dans des modèles in vitro [60] et in vivo chez des rongeurs [58, 59] sont encourageants, et à notre connaissance, seule une de ces pistes thérapeutiques a atteint le stade clinique (BIIB094, NCT03976349) (Figure 3, Tableau 1).
Par ailleurs, il a été démontré que dans la MP idiopathique, c’est à dire sans variant pathogène de LRRK2 ou d’un autre gène connu, une augmentation de l’activité kinase de la protéine LRRK2 est également présente [61]. Ainsi, ces thérapies ciblant LRRK2 pour les MP-LRRK2, si elles s’avéraient efficaces, pourraient avoir un effet bénéfique dans la MP idiopathique [51].
5. PRKN
Les variants bialléliques du gène PRKN sont la cause principale des MP de début précoce (âge moyen de début entre 25 et 35 ans). Le phénotype est assez spécifique, avec une bonne réponse à la L-dopa, une évolution lente, des dyskinésies et/ou des fluctuations motrices importantes, peu de troubles cognitifs et de symptômes non moteurs [9]. La protéine Parkin est une ubiquitine ligase E3. En association avec la kinase PINK1 (dont les variants bialléliques sont aussi responsables d’une forme précoce de MP), elle joue un rôle crucial dans la mitophagie en initiant la dégradation des mitochondries dysfonctionnelles [62]. L’accumulation d’espèces réactives à l’oxygène (ROS) étant le principal déclencheur de la mitophagie, le dysfonctionnement de ce mécanisme de dégradation aboutit à des niveaux anormalement élevés de ROS qui deviennent cytotoxiques, en particulier vis-à-vis des neurones dopaminergiques [63]. Au-delà de la perte de la fonction mitophagique de Parkin, sa diminution d’expression augmente la vulnérabilité des neurones dopaminergiques à la neuro-inflammation [64]. Parkin a de plus un rôle neuroprotecteur contre la toxicité de l’alpha-synucléine [65].
À ce stade, aucun traitement qui cible spécifiquement et directement la Parkin ou son gène n’a atteint le stade d’essai clinique. Néanmoins, de multiples petites molécules sont en phase préclinique, avec pour objectif principal de rétablir la fonction mitophagique. L’une des approches en cours d’évaluation est l’activation directe de la Parkin par l’altération des mécanismes d’auto-inhibitions. En effet, en condition basale, la Parkin adopte une conformation autoinhibitrice qui peut être ciblée et modifiée par certaines molécules [66]. Les premiers essais ont montré cet effet in vitro [66], mais il reste à confirmer in vivo. Une autre stratégie thérapeutique visant au rétablissement de la fonction mitophagique de la Parkin est l’inhibition du système ubiquitine protéasome 30S dont le rôle à l’état physiologique est de cliver l’ubiquitine des substrats de Parkin et ainsi contrebalancer l’action mitophagique médiée par la Parkin [67]. L’inhibition de cette enzyme pourrait potentiellement compenser la réduction de l’activité mitophagique de la Parkin. Deux composés (MF-094 et MF-095) ont montré une inhibition efficace et sélective de l’ubiquitine protéasome 30S in vitro [68]. Ces stratégies ont pour objectif d’améliorer l’efficience de la protéine Parkin produite.
Théoriquement, pour les variants conduisant à une perte de fonction, il serait possible d’exprimer la Parkin qui fait défaut par des approches de thérapie génique par exemple. Néanmoins, un certain nombre d’obstacles (perte neuronale déjà massive, difficulté à cibler les neurones dysfonctionnels et à contrôler précisément le niveau d’expression de la Parkin, etc.) rendraient difficile sa mise en œuvre. Aucune approche de thérapie génique n’est actuellement à l’essai pour PRKN.
6. Challenges, stratégies et directions futures
Depuis la découverte de l’implication du gène SNCA dans la MP en 1997, de nombreux gènes responsables de formes monogéniques ont été décrits [6, 69]. Ces découvertes ont fourni de nouvelles connaissances sur les mécanismes biologiques sous-jacents de la maladie et ont été intégrées dans la stratégie diagnostique mise en œuvre devant des formes précoces et/ou familiales. Dès lors, la stratification des patients selon leurs variants génétiques est possible, ouvrant la voie aux essais thérapeutiques ciblant des gènes, voire des variants précis [70]. Alors que de nombreux essais précliniques sont en cours, les essais cliniques sont plus rares et ne concernent qu’un nombre limité de gènes (SNCA, GBA1 et LRRK2), et aucun n’a encore franchi le stade d’une approbation par une agence du médicament.
En effet, plusieurs difficultés coexistent :
- Les formes génétiques sont individuellement assez rares d’où la difficulté de constituer des cohortes suffisamment grandes, étape nécessaire pour atteindre une puissance statistique suffisante. C’est surtout vrai pour SNCA, un peu moins pour LRRK2 et GBA1 dont les mutations sont plus fréquentes. Dans cette perspective, les collaborations internationales, la création de registres de données longitudinales ainsi que leur partage sont précieux et indispensables pour la réalisation d’essais cliniques d’envergure.
- L’évaluation de l’efficacité d’un traitement est difficile dans une maladie d’évolution relativement lente. Ce dernier point est d’autant plus vrai pour les MP-PRKN qui ont une évolution s’étalant généralement sur de nombreuses décennies. Il est important de pouvoir connaître l’histoire naturelle de ces formes génétiques de MP et de les modéliser à l’échelle individuelle.
- L’absence de biomarqueurs complique également l’évaluation de l’efficacité. La vérification de l’engagement de la cible au niveau biologique n’est pas encore possible pour SNCA, GBA1 et PRKN. Néanmoins, pour LRRK2, il a été montré que le taux urinaire de RAB10 phosphorylé sur la thréonine 73 est associée positivement à la progression de la MP, permettant de suivre partiellement l’activité LRRK2 [71, 72]. Certains tests récemment développés permettent également de mesurer le niveau et l’activité de LRRK2 dans le sang [73].
- Certaines molécules ne passent pas la barrière hémato-encéphalique (BHE) et ainsi n’atteignent pas leur cible biologique. Des équipes de recherche élaborent de nouvelles stratégies pour augmenter le passage de la barrière hémato-encéphalique, notamment par l’utilisation de nouveaux récepteurs, de nouveaux transporteurs, d’ultrasons ou encore par la création de nanomatériaux [74, 75].
- L’identification du moment opportun pour débuter le traitement dans une maladie où les mutations génétiques s’expriment dès le développement du système nerveux donc bien avant le début des signes cliniques reste difficile.
- La dégénérescence neuronale débute au moins une dizaine d’années avant le début des symptômes cliniques. L’utilisation de traitements pendant cette phase présymptomatique pourrait donc à l’avenir permettre de retarder, voire de prévenir, la survenue de la maladie. L’identification des individus à risque, notamment via la génétique, serait alors essentielle pour débuter un traitement en phase présymptomatique. Néanmoins, rappelons que la majorité des individus porteurs de variants à risque LRRK2 et GBA1 ne développeront pas la maladie malgré un risque largement accru comparé au reste de la population générale [15, 16]. Ainsi, comment déterminer quel individu à risque développera la maladie et donc lequel traiter de manière présymptomatique ? La réponse à cette question est complexe et dépasse le cadre de cette revue. Il est probable que, chez les individus porteurs à risque, la prise en compte de biomarqueurs et de données cliniques, associés aux données génétiques et environnementales, sera nécessaire pour distinguer ceux qui développeront la maladie des autres et pour choisir la meilleure fenêtre d’intervention.
- L’administration du traitement doit être simple, sécurisée et acceptable pour les patients, car elle sera probablement prolongée du fait de l’évolution relativement lente de la maladie.
Néanmoins, les progrès réalisés ces dernières décennies montrent qu’il est possible d’espérer que la MP entre dans l’ère de la médecine de précision grâce aux connaissances générées par les découvertes faites dans les formes monogéniques et à la possibilité de stratifier les patients selon leur cause génétique. La question majeure sera alors celle de l’applicabilité de ces thérapies ciblées aux autres patients atteints de MP idiopathique. Cette avancée majeure pourrait changer complètement l’évolution de cette maladie qui, plus de 200 ans après sa découverte, reste à ce jour incurable [76].
Déclaration d’intérêts
Les auteurs ne travaillent pas, ne conseillent pas, ne possèdent pas de parts, ne reçoivent pas de fonds d’une organisation qui pourrait tirer profit de cet article, et n’ont déclaré aucune autre affiliation que leurs organismes de recherche.