


 CC-BY 4.0
CC-BY 4.0
1. Setting the scene: allostery and the lactose repressor
Back in 1961, in the parent of this series at the French Academy of Sciences, Jacob and Monod published the model of the operon. Worth noticing, this outstanding work appeared in French [1]. This model provided biology with a universal concept explaining how the expression of genes responds to environmental or local cues, so that cells accommodate inevitable variations of their internal or external state. In detailed extensions of the model, a genetically encoded regulator was postulated to control the transcription of a gene succession into an intermediary messenger RNA molecule (mRNA) used as a template for the synthesis of proteins belonging to the regulatory unit. The concept abstracted experimental observations made in the commensal bacterium Escherichia coli when it uses lactose as its carbon source. The proposed regulatory scheme allowed the cell to adjust the carbon demands of its metabolism to its environmental supply via the use of a regulator.
It was subsequently found that the regulator, LacI, was a protein made of four identical subunits [2]. The LacI tetramer displayed two stable conformations, depending on the presence of allolactose—an isomer of lactose—in the cell’s cytoplasm. In the absence of this inducer molecule—or of a chemical analog such as isopropyl-𝛽-D-thiogalactoside, IPTG—the regulator was binding to the DNA region upstream of the genes of interest, named the operator. The set of gene sequences regulated by LacI constituted the lactose operon. In the absence of IPTG, LacI sticked to the operator and prevented RNA polymerase to begin transcription of the cognate transcription unit into an mRNA. Conversely, when bound to IPTG, another form of LacI was unable to bind the operator, allowing transcription to begin. Briefly, LacI acts as a repressor, a molecular switch responsive to IPTG, with operon activity OFF in the absence of the inducer and ON in its presence. The deep question which is still under investigation today is how LacI can undergo the considerable conformational change that oscillates between a structure with affinity for the operator-DNA and another structure, open for IPTG binding and without operator affinity.
The OFF/ON transcription capability of LacI to bind to an operator region, via blocking and releasing RNA polymerase for transcription initiated from a promoter is routinely used in Synthetic Biology constructs to regulate gene expression. Understanding the dynamics of this transition and being able to engineer variants of LacI or similar repressors with finely tuned properties is obviously a valuable engineering goal. Based on genetic, physiologic and biochemical experiments, an influential model proposed in 1965 by Monod, Wyman and Changeux (MWC) showed how a pre-existing equilibrium between two forms of a protein complex would account for a switch-like behaviour [3]. Yet the model, involving so-called “allosteric transitions”, did not come up with a detailed molecular mechanism allowing us to understand how the equilibrium is achieved and displaced upon binding of an effector molecule of interest. Furthermore, the model was based on the assumption that allosteric transitions were linked to symmetry properties, best illustrated in multi-subunit proteins, but superfluous if we try to understand these transitions. In fact, the molecular details of the corresponding process go far beyond standard allosteric transitions [4] but may even affect the way proteins are folded into three-dimensional shapes [5]. This conversion from one form to another also implies a dynamic whose evolution over time must be understood.
In this context, it is relevant to bear in mind that the thermal vibrations of atoms and amino acid residues in a protein at 300 K extend over the femtoseconds to picoseconds range [6]. In contrast, we expect that the protein movements pertinent to the transition will occupy the milliseconds range or be slower, hence are fit to embed the memory and recall of environmental events without being affected by the thermal background. The relevance of this time range is not unexpected because even longer time frames are involved in some catalytic reactions [7] and of course in processes leading to protein ageing via omnipresent chemical changes such as aspartate and asparagine isomerization [8]. Over the years, a considerable number of experiments investigated the role of mutations in the lacI gene, trying to understand how they would account for the allosteric transition. Despite more than five decades of research, the details regarding allosteric communication in this canonical molecular-switch are still being developed. A study of exceptional interest has just explored this time window using a mutation saturation experiment of the lacI gene with a study that produced no less than 60,000 mutations in the gene [9]. Remarkably, this study allowed the authors to uncover a completely unexpected type of regulatory behaviour of some of the LacI mutants, the ability to act as a band-pass (OFF/ON/OFF) or band-stop (ON/OFF/ON) filter. This observation opens up new interpretations of the physico-chemical background responsible for triggering the allosteric transition. It also prepares for novel designs for the regulation of Synthetic Biology constructs.
2. Regulation of transcription: an embodiment of a selective stabilization process
To visualize how this study allows us to better understand the transition pathways fixing how a protein or a protein complex may explore different conformations, let us go back to what we know of the LacI shape transitions, in formal terms. In this endeavour and as should be usual, it is critical to distinguish between observations and interpretations of the genotype/phenotype landscape of the objects of interest, noting that interpretations are consistently plagued by anthropocentric assumptions. Allosteric transitions are phenomenologically concomitant with the binding of a particular effector compound (IPTG for example) at a site distant from the active site of the protein (in the case of LacI, its operator binding site) when the equilibrium between the OFF/ON states of the repressor is shifted from the operon transcription control OFF state to the ON state. Oftentimes, this is inaccurately described as the result of a directed action of the effector that is supposed to induce a change in the shape of the regulator, as would do an agent endowed with a will.
Yet, we must refrain from calling for magic instructive designs, giving the effector molecule some magical power to enforce a change in the shape of the protein. An unbiased way to understand how the regulation develops is to assume that, at the onset of the observation and in the absence of the inducer, there is a number—usually quite small—of ON forms, prone to bind IPTG, in a dynamic equilibrium with OFF forms. The equilibrium between forms, resulting from thermal fluctuations—significant at 300 K—predates the binding of the effector. When the inducer is present, its binding ensures selective stabilization of the protein into its ON form, which is unable to bind the lactose operon operator, while the effector is unable to bind to the OFF form. In a two-states situation, this binding pulls the protein complexes having the form that bind the effector out of the equilibrium mixture of forms. Lacking free ON forms, the mixture re-equilibrates under the constraints of thermal fluctuations, leading to a progressively vanishing amount of the repressing OFF form as the amount of the inducer increases, matching its affinity for the effector. The speed of the process will underlie that of the allosteric transition.
Four LacI wild type and mutant regulatory competences. The wild type behaviour is displayed in blue. In the absence of the LacI inducer IPTG (I) LacI binds the lactose operon operator and prevent transcription. Upon inducer binding transcription can proceed (RNA polymerase is shown in light blue). In green the symmetrical regulation operates: transcription proceeds in the absence of I, while I acts as a co-repressor. Two unexpected behaviours are also displayed in the figure. LacI can work as a band-pass filter (black) with repression in the absence or high concentration of I, while, at an intermediate concentration LacI does not bind to the operator, allowing transcription to proceed. Conversely, mutants of LacI behaved as a band-stop filter (red), with transcription allowed except for an intermediate concentration of I, where the repressor has an intermediate conformation that binds to the operator.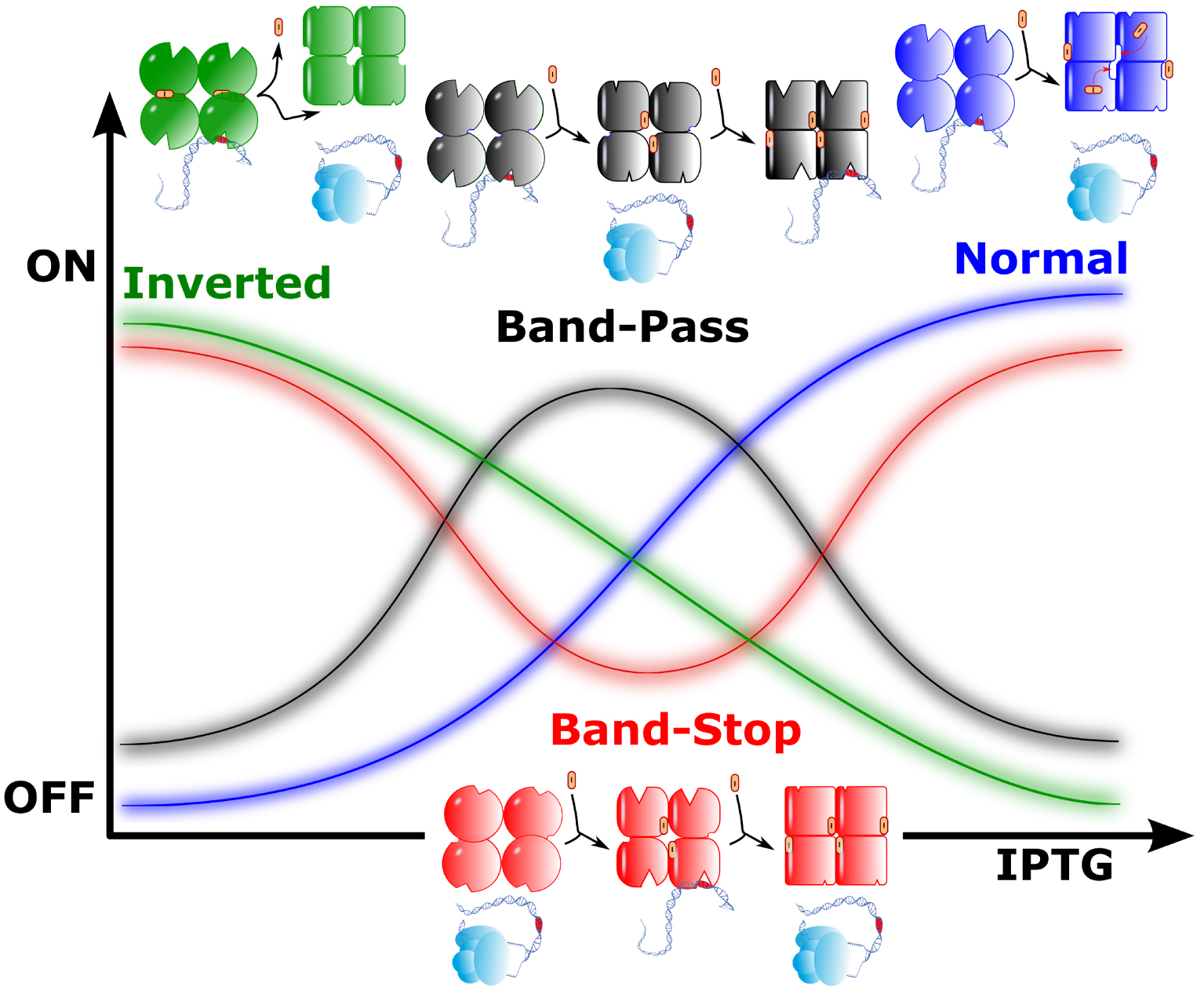
3. Myriads of mutations in the LacI polypeptide
Using an error-prone PCR, Tack et al. generated 62,472 lacI mutants. This remarkable prowess, represents an average of 7 nucleotide change per individual gene. Because 20 amino acids are encoded in 61 nucleotide triplets (codons), this still resulted on average in more than 4 simultaneous amino acid change per LacI mutant protein. Analysis of these mutants required a remarkable automation procedure, based on the expression of the lacZ gene, which is controlled by LacI. The highly involved experimental setup, not discussed here, compared expression of the mutants in a variety of environments [9]. This work allowed the authors to delineate the quantitative activation/inactivation pattern of each LacI mutant when responding to the inducer IPTG.
The wild type response can be visualized as a sigmoid curve, a raw concrete implementation of a logic “step function”. A number of the mutants displayed a similar curve, albeit with varying sensitivity to the presence of IPTG. This demonstrated that changes in many residues still retained the overall properties of the protein (Figure 1, Blue). In contrast, as could be generally anticipated, a large number no longer responded to IPTG, indicating that the mutant is no longer functional. However, analysis of some of the corresponding mutations allowed the authors to highlight amino acid residues that are involved in the binding of the targets or in the allosteric transition (not illustrated). Finally, another category of mutants revealed an inversion of the sigmoid curve, making IPTG a co-repressor rather than an inducer (Figure 1, Green). This mutant phenotype has commonly been observed previously in related repressors [10]. Both categories of LacI regulators could be predicted by the MWC model. Because most mutants comprised several mutations simultaneously, this work further uncovered an important feature: some mutations which led to an inactive form of the repressor could be compensated by another mutation located elsewhere in the protein sequence, restoring its activity. This co-variation, discussed below, implies some kind of interaction between the relevant residues. Interestingly, many were not close together in the polypeptide sequence.
This work also brought forth a surprise: a very important and unexpected family of regulators was uncovered in the collection of mutants. Some 200 mutants of LacI (among several thousands) behaved as “band-pass” (OFF/ON/OFF) or “band-stop” (ON/OFF/ON) filter switches (Figure 1, Black and Red, respectively). Remarkably, these novel activities evolved from combinations of amino acid substitutions that were almost silent when isolated. This behaviour is not predicted by the MWC model. In order for it to be understood we must take into account the actual molecular processes that make the allosteric transition develop concretely, in space and time. To be sure, the very fact that a state transition could be reversed in the protein complex when progressively increasing the concentration of an effector implies that some molecular event involving the effector happens during the course of the transition as it unfolds. This suggests that a significant piece of the protein changes its conformation during the transition, transiently opening up possible interactions with compounds such as the effector, if present in the environment. This potential for opening up interactions with the surroundings of a protein is a signature of the transitions that affect so-called “intrinsically disordered” protein regions, leading them to “phase transitions” [11]. In this context, disordered regions have indeed been shown to be critical in allosteric transitions [12]. In the case of LacI, the situation is particularly complex because the protein is a tetramer, so that intermediary states may necessitate transient association of subunits into dimers. This would uncover states that could bind the effector for a time and then channel the protein to a particular conformation, depending on the availability of the effector in a narrow concentration range, for example.
4. The role of protein “sectors”
A great many experimental studies and dynamic modelling of proteins have explored this type of situation, trying to highlight the amino acid residues of the protein involved in the transition. This is beautifully illustrated in the work of Ranganathan and co-workers. These authors identified protein regions they named “sectors” that were critical to allow for the transition [13]. In a variety of approaches, these and following works established that a large fraction of the amino acid residues of the protein did not belong to these sectors and could, remarkably, be changed into almost any of the 20 residues that make proteins without considerable changes in the protein activity [14]. In contrast, a network of specific residues remains critical to generate a functional protein structure. This is easily understood: if the nature of the amino acid residues were indifferent, two proteins with any sequence of the same size could be folded into widely different enzyme, a ribonuclease or a protease, for example. Furthermore, the need for specific residues does not only apply for catalysis but for allowing allosteric transitions as well. To be sure, among important families of functional residues, allosteric transition-related residues (a small number of positions in a given protein) form co-evolving networks throughout the structure—i.e. producing architectures prone to mediate long-range communication in the protein. A hallmark of these allosteric positions is an extreme sensitivity to perturbation. This requirement explains why, besides catalytic residues, the residues making sectors have a critical role and cannot be mutated freely, as indeed observed in the LacI experiment [9].
In summary, a change in a protein that led to its inactivation will often open up the possibility for a compensating change in one or several other positions of the sector to restore activity [15]. Do we have indications of the way these associated residues are organized in the protein? Remarkably, the key observation made by experiments following Ranganathan’s work was that sectors, when displayed in three-dimensional structures, corresponded to contiguous regions, while they were rarely contiguous in the primary sequence of the protein. This demonstrated that some sort of physico-chemical process, not directly derived from the protein backbone, ensures continuity between relevant residues. Figure 2 illustrates the simplified case of a protein made of a single subunit with two conformations. Mutations that are connected to allosteric transitions form specific networks (sectors) of amino acids within the protein structure (blue spheres) to influence active site function from a distance (red arrows) as a function of the presence of an effector molecule.
Allosteric transition in a monomeric protein. Amino acid residues are represented as spheres. Blue spheres label sectors that are involved in the allosteric transition and in catalysis. Binding of an effector (E) stabilizes the inactive form, while dissociation of the effector allows catalysis to proceed on a substrate (S). The red arrows show how sectors connect the catalytic site to the effector site, Movement of the polypeptide chain is triggered by connection between the residues and polypeptidic backbone presumably via a network of hydrogen bonds involving water molecules associated to the protein (see Figure 3).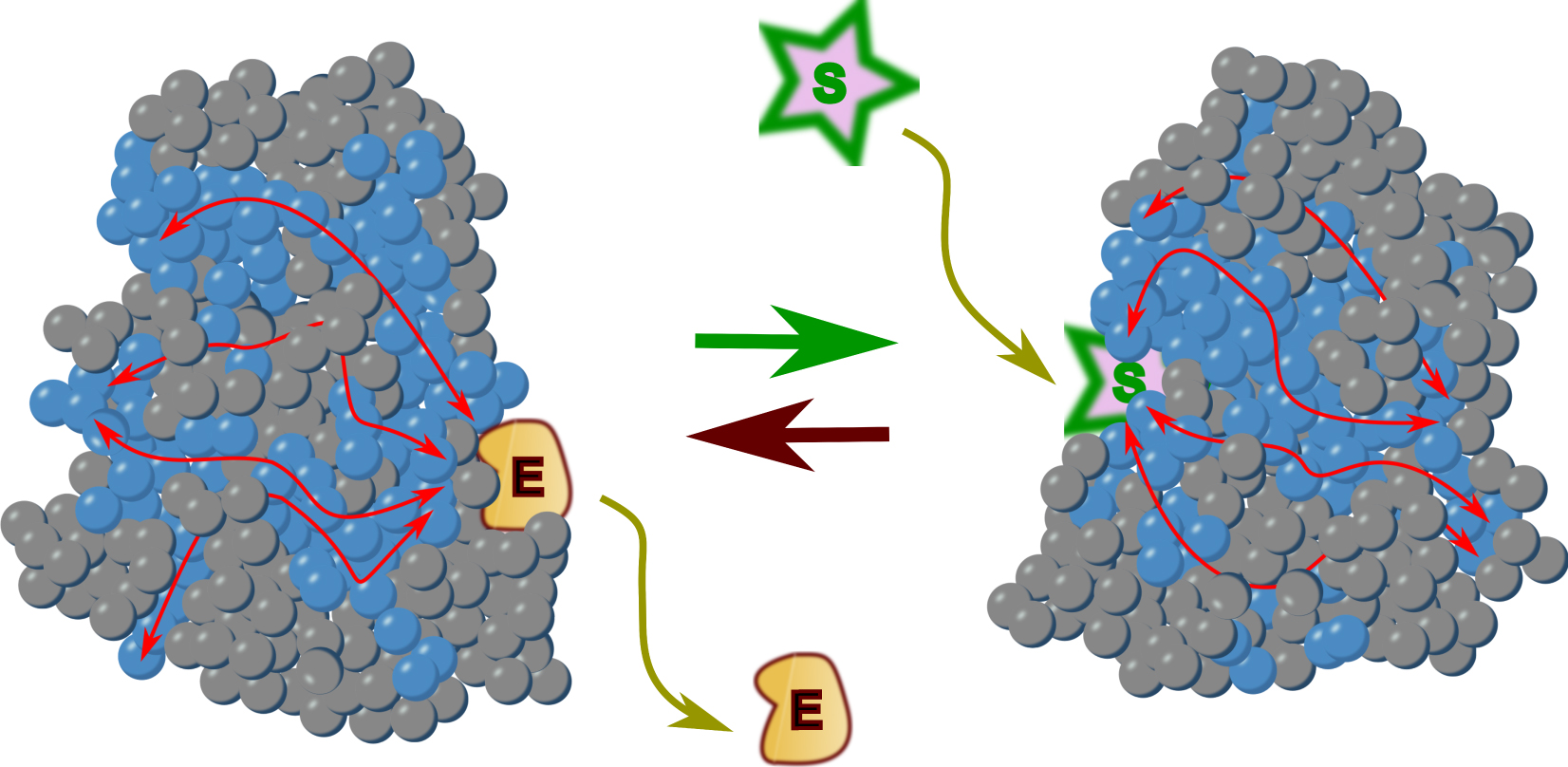
Here, sectors are identified as co-evolving units of protein structures associated with functional properties of proteins of interest—catalysis, binding, and allosteric regulation. In the case of allostery, a sector residue could sometimes be replaced by another one without significantly altering the behaviour of the protein, but this will have the consequence that another amino acid in the sector may cause the allosteric behaviour to change to a completely different behaviour in a subsequent mutation. This “latent” information explains why the band pass/band-stop phenotype revealed that it is covariant mutations that generate a cooperative internal architecture within the protein structure. The corresponding amino acid residues form specific amino acid arrays within the protein structure and remotely influence the function of the active site. This behaviour is consistent with a wire-like architecture of sectors, connecting sites along a three-dimensional pathway including some distantly positioned residues at the expense of some more proximal ones [16]. At this point in the interpretation of the data it becomes interesting to note that the sectors involved in allosteric transitions are not only found in proteins, they also characterise functional RNA molecules [17]. This new observation suggests a critical rationale for this process: a general property associated with very different biological polymers, proteins and nucleic acids, must be proposed as being able to link their functional properties and structure. The question then arises as to what might be the source of this ubiquitous property.
5. Water and hydrogen bonds as the connecting process
Many paths may account for the formation of sectors. A seldom explored idea would involve water in the process. Water is the universal support of life, and perhaps for this reason, it is more or less “invisible” in structural studies. Most X-rays diffraction analyses present proteins or nucleic acids as if standing in an empty medium, with a few water molecules located here and there. Yet, water’s role goes far beyond its role as a solvent or its dipole moment. Water in contact with a protein has its protons organised along a network of hydrogen bonds (Figure 3). This structural feature is visible in proton resonance identified in NMR studies [18]. The network of protons surrounding the macromolecules and engaged in hydrogen bonds is strongly reminiscent of the possible states of an “Ising model” (see [19], as an example of many instances of the question in biology). In the Ising model, each lattice site only interacts with the sites directly adjacent to it on the lattice, while being affected by a general field in which the compound of interest is embedded.
An illustration of the presence of identified water molecules in a protein crystal. Homo sapiens protein-tyrosine phosphatase 1B has been crystallized at 1.95 Å resolution (PDB 1SUG entry, https://www.rcsb.org/3d-view/1SUG/). The polypeptide backbone is illustrated and water molecules that could be identified in the crystal are shown as red dots. It can be seen on the figure that the distribution of these molecules is not random and follows the fold of the protein.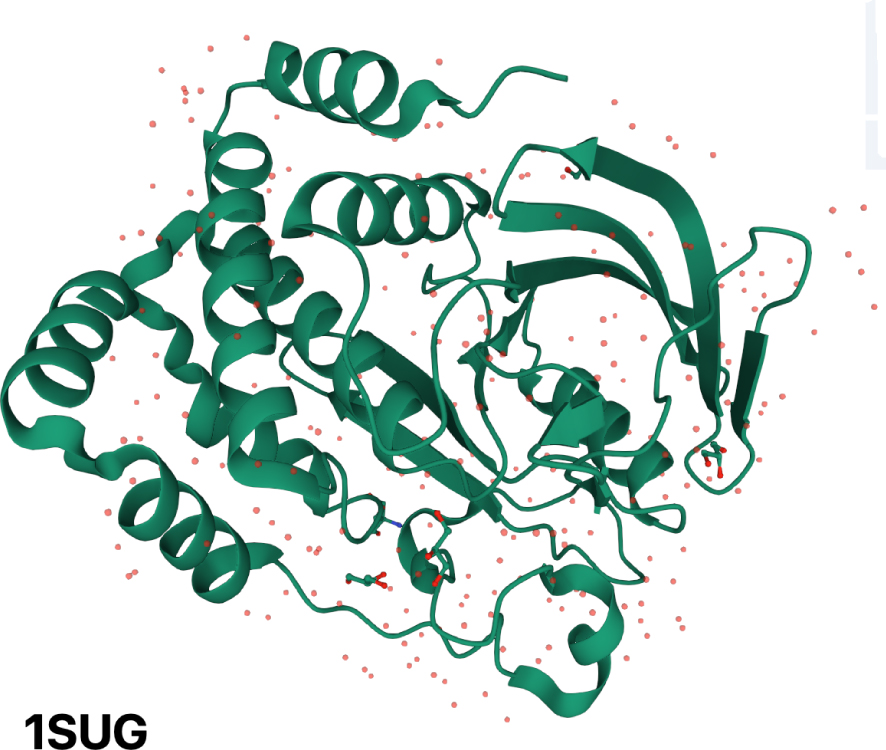
At 300 K and in a particular cellular environment, there is an equilibrium between the possible states of hydrogen bonds constrained by a macromolecule, and the dominant states are generally strongly shifted in the direction of stabilizing a particular 3D structure. Now, hydrogen bonds can move from one acceptor to another very quickly (the energy involved is of the order of thermal energy). This can happen in a concerted way, through interactions with specific amino acids of the biopolymer, but also with the polypeptide backbone, moving from one 3D structure to another (typically the MWC situation described in 1965), depending on the presence of suitable substrates in the environment. In the course of the transition there must be a path followed by hydrogen bonds forming and breaking in a time sequence, likely to follow locally the chain of amino acid residues in the protein of interest. This must happen fast, but because it must develop sequentially in a “domino effect” [20] it must operate in a time span considerably much slower than most vibrations of the atoms making the protein (milliseconds to seconds for the exchange rate of a water molecule). In physico-chemical terms and analogous to the transition from one Ising state to another Ising state, this switching can be coupled with a mechanical change illustrated by a transition through flexible regions of the protein, with eventual re-equilibration of hydrogen bonds at greater distances, mainly within water, away from the protein’s backbone and amino acids.
In this context, we can propose here that the role of the sectors is to maintain a local organisation of the water molecules such that they form a network of hydrogen bonds, both between the water molecules and the protein and between water molecules. This corresponds to the families of LacI mutants and their transitions. The authors of the study illustrated above did not propose a physico-chemical explanation for their observation, but it is most likely associated with the interaction of water with the protein, in a scenario similar to that of the proton “wires” identified in the respiratory complex [21]. The originality of their “U-type” filters can then be explained by a sequential change of a hydrogen bonds network that reverses or bifurcates to another structure when the effector concentration increases.
The authors of the LacI study noted that this behaviour is related to some “disorder” in the local sequence of the protein, a feature that is directly derived from the local structure of water. We have just discussed how allosteric transitions require the presence of an unstable transient state of a part of the allosteric protein involved. The nature of this disorder—which would be better termed “flexibility” rather than disorder, since the very concept of order involves hidden information provided by the observer—has certainly something to do with the local reorganisation of water. This type of transition temporarily opens the possibility for water molecules to form transient hydrogen bonds with the polypeptide backbone or specific amino acid side chains. As a result, the dynamics of entropic changes related to the water structure can play an important role in explaining a phenotype such as a band-pass or band-stop filter. When a ligand reaches a binding site in the protein, it exchanges for a few molecules of water, triggering a chain of hydrogen bond breakage and formation that will initiate a transition from the initial state of the protein to another state. It should be noted that, triggered by thermal flucutations, this transition can also exist in the absence of a ligand, possibly strongly displaced in one direction rather than another. The role of the ligand will then be to select a particular state, so that the equilibrium is shifted until all possible binding sites are occupied by the ligand (this is what is described in the MWC model and discussed at the beginning of this text). If one accepts this view, there is no “instruction” from the ligand to tell the protein to change shape, but selection of a possibly minor shape, followed by re-equilibration of the mixture.
6. Yet another look at water: as an information support allowing memorization and recall, water must only have very short memory
A final note. For sociological reasons that should be explored, water, which is an element so widespread that we forget its highly original properties—note that its volume increases when it freezes—has been the subject of much magic thinking, in particular by people with little interest in scientific knowledge. The far-fetched idea that this solvent could carry the long term memory of past events has been an excuse for Britons to laugh at the gullibility of the French mass media [22]. Yet, there is a link between water and memory, but its role is exactly the opposite of that of a long-term memory, since water’s memory is subject to rapid erasure. Water acts as a medium to carry and erase very quickly the memory of its past states, as a general carrier of the hydrogen bond networks that are used to unobtrusibly carry biological information. Seen in this way, water enables cells to compute, just as electrons do in electronic devices. With this remark we wish to highlight the crucial role of water in biological studies, making analyses that represent biological objects as if they were in an empty space condemned to miss one of the most important properties of life.
French version
1. Plantons le décor : l’allostérie et le répresseur de l’opéron lactose
En 1961, dans ces mêmes Comptes Rendus des Séances de l’Académie des Sciences, Jacob et Monod publiaient le modèle de l’opéron. Et il faut noter que ce travail remarquable était publié en français [1]. Ce modèle apportait à la biologie un concept universel expliquant comment l’expression des gènes répond à des signaux environnementaux ou endogènes, de sorte que les cellules s’adaptent aux variations inévitables de leur état interne ou externe. Des extensions détaillées du modèle postulaient qu’un régulateur génétiquement codé contrôlait la transcription d’une succession de gènes en une molécule intermédiaire d’ARN messager, utilisé comme matrice pour la synthèse des protéines appartenant à l’unité de régulation. Ce concept universel s’inspire d’observations expérimentales faites chez la bactérie commensale Escherichia coli lorsqu’elle utilise le lactose comme source de carbone. Le schéma de régulation proposé permet de comprendre comment la cellule, par le truchement d’un régulateur, ajuste les besoins en carbone de son métabolisme à ce qu’offre son environnement.
Par la suite, il a été découvert que le régulateur était une protéine composée de quatre sous-unités identiques [2]. Ce tétramère, LacI, se présente sous deux formes stables en fonction de la présence d’allolactose — un isomère du lactose — dans le cytoplasme de la cellule. En l’absence de cet inducteur — ou d’un analogue synthétique comme l’isopropyl-𝛽-D-thiogalactoside, IPTG — le régulateur se lie à une région de l’ADN en amont des gènes de transport et de dégradation du lactose, appelée opérateur. L’ensemble des séquences des gènes régulés par LacI constitue l’opéron lactose. En l’absence d’IPTG, LacI attaché à l’opérateur empêche l’ARN polymérase de commencer la transcription. Inversement, lorsqu’il est lié à l’IPTG, LacI, sous une forme différente, est incapable de se fixer à l’opérateur, et la transcription commence. En bref, LacI agit comme un répresseur, un interrupteur moléculaire sensible à l’IPTG, l’activité de l’opéron étant désactivée en l’absence de l’inducteur et activée en sa présence. La question profonde qui est toujours en cours d’investigation aujourd’hui est de savoir comment LacI peut connaître le changement conformationnel considérable qui oscille entre une structure qui a de l’affinité pour l’opérateur/ADN et une autre structure, permettant à l’IPTG de s’y lier, et sans affinité pour l’opérateur.
La capacité de LacI à se lier à un opérateur et à contrôler la transcription de gènes choisis (états INACTIF/ACTIF), via le blocage et la libération de l’ARN polymérase à partir d’un promoteur est couramment utilisée dans les constructions de biologie synthétique destinées à réguler leur expression. Comprendre la dynamique de cette transition et savoir créer des variants de LacI ou de répresseurs semblables en affinant leurs propriétés est évidemment un objectif d’ingénierie important. Fondé sur des expériences génétiques, physiologiques et biochimiques, un célèbre modèle proposé en 1965 par Monod, Wyman et Changeux (MWC) montre comment un équilibre préexistant entre deux formes d’un complexe protéique peut expliquer un comportement de type interrupteur [3]. Cependant, ce modèle, qui fait intervenir des « transitions allostériques », n’explicite pas le mécanisme moléculaire détaillé qui permettrait de comprendre comment l’équilibre est atteint et déplacé lors de la liaison d’une molécule effectrice. Au surplus, ce modèle repose sur l’hypothèse que les transitions allostériques sont associées à des propriétés de symétrie, bien illustrées chez les protéines multi-sous-unités, mais en réalité superflues pour comprendre ces transitions. De fait, les détails moléculaires des processus correspondants vont bien au-delà des transitions allostériques standard [4] et peuvent même affecter la manière dont une protéine se replie dans l’espace [5]. Cette conversion d’une forme à une autre implique aussi une dynamique dont il faut comprendre l’évolution dans le temps.
Dans ce contexte, il est important de garder à l’esprit que les vibrations thermiques des atomes et des acides aminés dans une protéine à 300 K vont de la femtoseconde à la picoseconde [6]. En revanche, nous nous attendons à ce que les mouvements de la protéine pertinents pour la transition soient de l’ordre de la milliseconde ou plus lents, et puissent donc prendre en compte la mémorisation et le rappel des événements qui affectent l’environnement sans être influencés par le bruit thermique. La pertinence de cette plage de temps n’est pas inattendue car des délais encore plus longs sont impliqués dans certaines réactions catalytiques [7] et bien sûr dans les processus conduisant au vieillissement des protéines via des changements chimiques omniprésents comme l’isomérisation de l’aspartate et de l’asparagine [8]. Au fil des ans, un nombre considérable d’expériences ont étudié le rôle des mutations du gène lacI, en essayant de comprendre comment elles pourraient expliquer la transition allostérique. Malgré plus de cinq décennies de recherche, les détails concernant la communication allostérique dans cet interrupteur moléculaire canonique sont toujours en cours. Une étude d’un intérêt exceptionnel vient d’explorer cette fenêtre temporelle en utilisant une expérience de saturation en mutations du gène lacI dans un travail qui a produit pas moins de 60 000 mutations dans le gène [9]. De façon remarquable, cette étude a permis aux auteurs de découvrir un type de comportement régulateur totalement inattendu chez certains des mutants LacI, la capacité d’agir comme filtre passe-bande (INACTIF/ACTIF/INACTIF) ou coupe-bande (ACTIF/INACTIF/ACTIF). Cette observation ouvre à de nouvelles interprétations le contexte physico-chimique responsable du déclenchement de la transition allostérique. Elle apporte aussi de nouvelles conceptions pour la construction de régulations utiles à la biologie synthétique.
2. Régulation de la transcription : concrétisation d’un processus de stabilisation sélective
Pour illustrer comment cette étude nous permet de mieux comprendre le chemin des transitions qui conduisent une protéine ou un complexe protéique à explorer différentes conformations, revenons à ce que nous savons des changements de forme de LacI, en termes formels. À cette fin, et comme on devrait toujours le faire, il faut bien faire la distinction entre les observations et les interprétations de la diversité des génotypes/phénotypes des objets en cause, en notant que les interprétations sont constamment entachées d’anthropocentrisme. Les transitions allostériques sont phénoménologiquement concomitantes de la liaison d’un effecteur particulier (IPTG par exemple) à un site distant du site actif de la protéine (dans le cas de LacI, son site de liaison à l’opérateur) lorsque l’équilibre entre les états INACTIF/ACTIF du répresseur est déplacé de l’état INACTIF du contrôle de la transcription de l’opéron à l’état ACTIF. Bien souvent, ce phénomène est décrit de manière inexacte comme le résultat d’une action dirigée de l’effecteur qui est censé induire, comme le ferait un objet animé d’une volonté, un changement de forme du régulateur.
Pourtant, nous devons nous abstenir de faire appel à des conceptions instructives, conférant à la molécule effectrice un pouvoir magique qui imposerait un changement de forme à la protéine. Une façon impartiale de comprendre comment la régulation se développe est de supposer qu’au début de l’observation et en l’absence de l’inducteur il existe un nombre — généralement faible — de protéines dans l’état ACTIF, enclines à fixer l’IPTG, en équilibre dynamique avec beaucoup d’autres dans l’état INACTIF. L’équilibre entre les formes, résultant des fluctuations thermiques, significatives à 300 K, précède la liaison de l’effecteur. Lorsque l’inducteur est présent, sa liaison assure la stabilisation sélective de la protéine dans son état ACTIF, incapable de lier l’opérateur de l’opéron lactose, tandis que cet effecteur est incapable de se lier à la forme qui est dans l’état INACTIF. Dans une situation à deux états, cette liaison extrait les complexes protéiques ayant la forme qui lie l’effecteur hors du mélange des formes en équilibre. Aussi, en l’absence de formes dans l’état ACTIF libres, le mélange se rééquilibre sous la contrainte des fluctuations thermiques, conduisant à une quantité progressivement décroissante de la forme dans l’état INACTIF répressive au fur et à mesure que la quantité d’inducteurs augmente, coïncidant avec son affinité pour l’effecteur. La vitesse du processus sous-tend celle de la transition allostérique.
3. Des myriades de mutations dans le polypeptide LacI
En utilisant une PCR infidèle, Tack et al. ont créé 62 472 mutants du gène lacI. Cette prouesse remarquable représente une moyenne de 7 changements de nucléotide par gène. Comme les 20 acides aminés sont codés par 61 triplets de nucléotides (codons), cela conduit en moyenne à plus de 4 changements simultanés d’acides aminés par protéine LacI mutante. L’analyse de ces mutants a impliqué une automatisation remarquable, fondée sur l’expression du gène lacZ contrôlé par LacI. Le dispositif expérimental très complexe, qui n’est pas discuté ici, a comparé l’expression des mutants dans un grand nombre d’environnements [9]. Ce travail a permis aux auteurs de construire un modèle quantitatif d’activation/inactivation pour chaque mutant LacI lorsqu’il répond à l’inducteur IPTG.
La réponse du type sauvage peut être décrite sous la forme d’une courbe sigmoïde, implémentation concrète grossière d’une « fonction échelon unité » logique. Un certain nombre de mutants ont conservé une réponse du même type, avec une sensibilité variable à la présence d’IPTG. Cela démontre que changer bien des acides aminés conserve malgré tout les propriétés globales de la protéine (Figure 1, Bleu). En revanche, comme on pouvait s’y attendre, un grand nombre de mutants ne répondent plus à l’IPTG, ce qui indique qu’ils ne sont plus fonctionnels. Cependant, l’analyse de certaines des mutations correspondantes a permis aux auteurs de mettre en évidence des acides aminés qui sont impliqués dans la liaison de l’effecteur ou de l’opérateur, ou encore dans la transition allostérique (non illustré). Enfin, une autre catégorie de mutants a révélé une inversion de la courbe sigmoïde, faisant de l’IPTG un corépresseur plutôt qu’un inducteur (Figure 1, Vert). Ce phénotype de mutant a été déjà communément observé chez des répresseurs apparentés [10]. Ces deux catégories de régulateurs de LacI sont d’un type prédit par le modèle MWC. Comme la plupart des mutants comprennent plusieurs mutations simultanément, ce travail a aussi permis de découvrir une caractéristique importante : certaines mutations qui conduisent à une forme inactive du répresseur peuvent être compensées par une autre mutation située ailleurs dans la séquence de la protéine, restaurant son activité. Cette co-variation, discutée plus bas, implique une certaine forme d’interaction entre les acides aminés concernés. Il est intéressant de noter que beaucoup d’entre eux ne sont pas proches les uns des autres dans la séquence polypeptidique.
Quatre formes de régulation associées à LacI de type sauvage et mutant. Le comportement du type sauvage est affiché en bleu. En l’absence de l’inducteur (IPTG, I), LacI se lie à l’opérateur de l’opéron lactose et empêche la transcription (bleu). Après la liaison de l’inducteur, la transcription peut se poursuivre (l’ARN polymérase est représentée en bleu clair). En vert, c’est une régulation symétrique qui opère : la transcription se déroule en l’absence de I, tandis que I agit comme un co-répresseur. Deux comportements inattendus sont également représentés sur la figure. LacI peut fonctionner comme un filtre passe-bande (noir) avec une répression en l’absence ou à une concentration élevée de I, alors qu’à une concentration intermédiaire, LacI ne se lie pas à l’opérateur, permettant à la transcription de se poursuivre. Inversement, les mutants de LacI se comportent comme un filtre coupe-bande (rouge), avec une transcription autorisée sauf pour une concentration intermédiaire de I, où le répresseur a une conformation intermédiaire qui se lie à l’opérateur.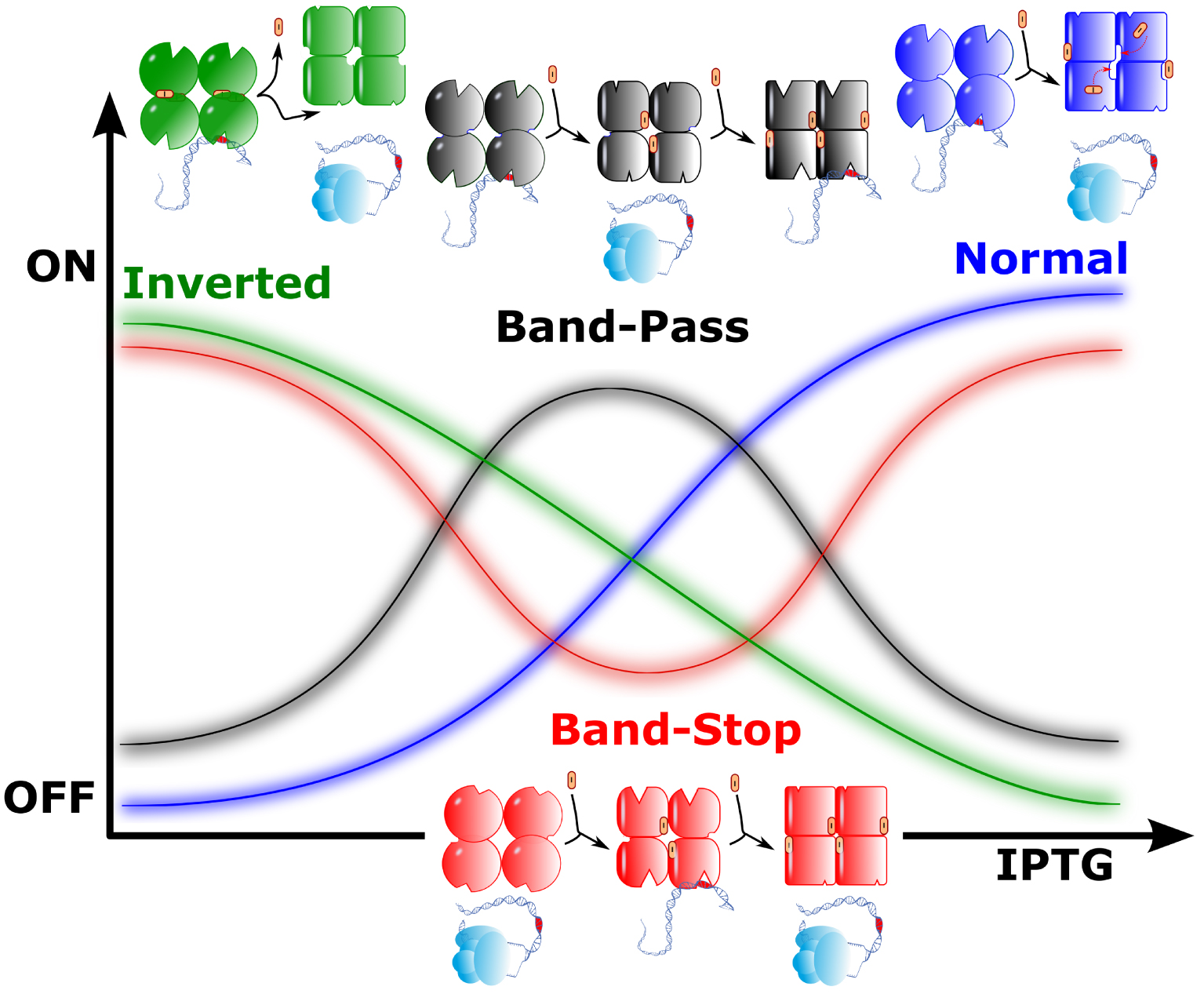
Ce travail apporte aussi une surprise : une famille de régulateurs très importante et inattendue a été ici mise au jour. Quelque 200 mutants de LacI (parmi plusieurs milliers) se comportent comme des filtres passe-bande (INACTIF/ACTIF/INACTIF) ou coupe-bande (ACTIF/INACTIF/ACTIF) (Figure 1, Noir et Rouge, respectivement). De manière remarquable, ces activités originales ont évolué à partir de combinaisons de substitutions d’acides aminés qui étaient presque silencieuses lorsqu’elles sont isolées. Ce comportement n’est pas prédit par le modèle MWC. Pour le comprendre, il nous faut prendre en compte les processus moléculaires réels qui font que la transition allostérique se développe concrètement dans l’espace et dans le temps. Le fait même qu’une transition d’un état à l’autre puisse être inversée dans le complexe protéique en parallèle avec l’augmentation progressive de la concentration d’un effecteur implique qu’un événement moléculaire impliquant l’effecteur se produise au cours du développement de la transition. Cela suggère qu’une partie importante de la protéine change de conformation au cours de la transition, et ouvre transitoirement des interactions possibles avec des composés comme l’effecteur, s’il est présent dans l’environnement. Cette dynamique d’ouverture d’interactions d’une protéine avec son environnement est une signature des transitions qui affectent les régions protéiques dites « intrinsèquement désordonnées », les conduisant à des « transitions de phase » [11]. Dans ce contexte, les régions désordonnées ont en effet été montrées comme critiques pour les transitions allostériques [12]. Dans le cas de LacI, la situation est particulièrement complexe car la protéine est un tétramère, de sorte que les états intermédiaires peuvent nécessiter l’association transitoire des sous-unités en dimères. Cela permettrait de mettre au jour des états qui pourraient lier l’effecteur pendant un certain temps et ensuite canaliser la protéine vers une conformation particulière, en fonction de la disponibilité de l’effecteur dans une gamme de concentration étroite, par exemple.
4. Le rôle de « secteurs » dans les protéines
De très nombreuses études expérimentales et de modélisation dynamique des protéines ont exploré ce type de situation, en essayant de mettre en évidence les acides aminés de la protéine impliqués dans la transition. Cela est magnifiquement illustré par les travaux de Ranganathan et de ses collaborateurs. Ces auteurs ont identifié des régions protéiques qu’ils ont nommées « secteurs », critiques pour permettre la transition [13]. Grâce à diverses approches, ces travaux et ceux qui ont suivi ont permis d’établir qu’une grande partie des acides aminés de la protéine n’appartenaient pas à ces secteurs et pouvaient, de manière peut-être étonnante, être remplacés par presque n’importe lequel des 20 acides aminés qui composent les protéines sans que l’activité de la protéine soit considérablement modifiée [14]. En revanche, un réseau d’acides aminés spécifiques reste essentiel pour parvenir à une structure protéique fonctionnelle. Cela se comprend aisément : si la nature des acides aminés était indifférente, deux protéines de même taille et de séquence quelconque pourraient être repliées sous la forme d’enzymes très différentes, une ribonucléase ou une protéase, par exemple. Au surplus, le besoin d’acides aminés spécifiques ne s’applique pas seulement à la catalyse, mais aussi aux transitions allostériques. De fait, parmi les familles importantes d’acides aminés fonctionnels, ceux qui sont liés aux transitions allostériques (un petit nombre de positions dans une protéine donnée) forment des réseaux unis par leur co-évolution dans toute la structure, montrant qu’ils produisent des architectures nécessaires à la médiation de la communication à longue distance dans les protéines. L’une des caractéristiques de ces positions allostériques est leur extrême sensibilité aux perturbations. Cette exigence explique pourquoi, outre les acides aminés catalytiques, les acides aminés formant les secteurs ont un rôle critique et ne peuvent être mutés librement, comme cela a bien été observé dans l’expérience LacI [9].
En résumé, un changement d’acide aminé dans une protéine ayant conduit à son inactivation ouvrira souvent la possibilité qu’un autre changement puisse compenser ce défaut dans une ou plusieurs positions du secteur et restaure ainsi l’activité perdue [15]. Avons-nous des indications sur la façon dont ces acides aminés associés sont organisés dans la protéine ? De façon remarquable, l’observation clé faite par les expériences qui ont suivi les travaux de Ranganathan est que les secteurs, lorsqu’ils sont représentés dans des structures tridimensionnelles, sont faits de régions contiguës, alors qu’ils sont rarement contigus dans la séquence primaire de la protéine. Cela démontre qu’un processus physico-chimique, non directement dérivé du squelette de la protéine, assure une forme de continuité entre les acides aminés pertinents. La Figure 2 illustre le cas simplifié d’une protéine constituée d’une seule sous-unité avec deux conformations. Les mutations qui sont liées aux transitions allostériques forment des réseaux spécifiques d’acides aminés (secteurs) au sein de la structure de la protéine (sphères bleues) et influencent la fonction du site actif à distance (flèches rouges) en fonction de la présence d’un effecteur.
Transition allostérique dans une protéine monomère. Les acides aminés sont représentés par des sphères. Les sphères bleues marquent les secteurs qui sont impliqués dans la transition allostérique et dans la catalyse. La liaison d’un effecteur (E) stabilise la forme inactive, tandis que la dissociation de l’effecteur permet la catalyse sur un substrat (S). Les flèches rouges montrent comment les secteurs relient le site catalytique au site effecteur. Le mouvement de la chaîne polypeptidique est déclenché par la connexion entre les acides aminés et le squelette polypeptidique vraisemblablement via un réseau de liaisons hydrogène impliquant des molécules d’eau associées à la protéine (voir Figure 3).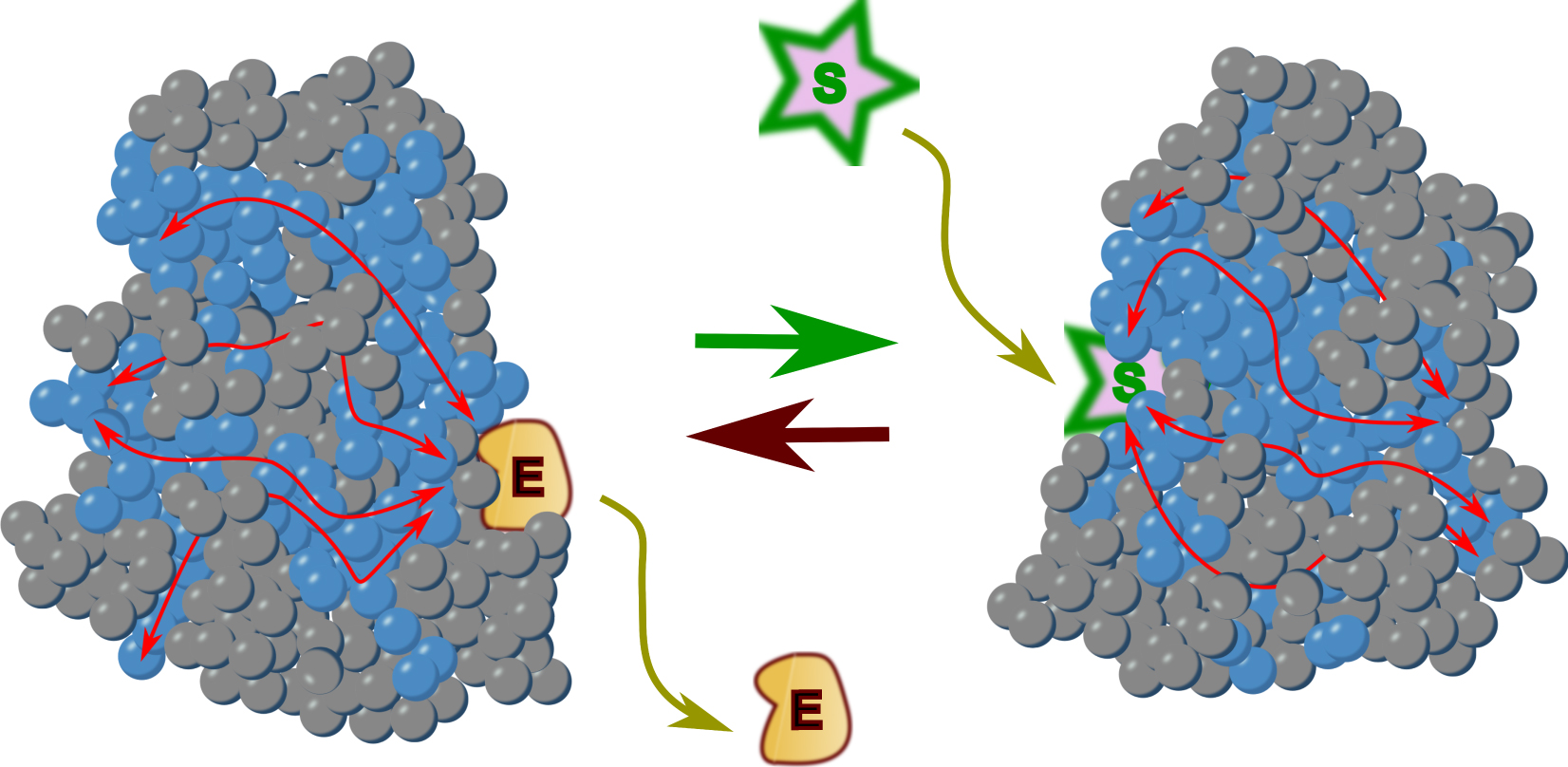
Ici, les secteurs sont identifiés comme des unités coévolutives des structures protéiques associées aux propriétés fonctionnelles de la protéine en cause, catalyse et régulation allostérique. Dans le cas de l’allostérie, un acide aminé d’un secteur peut parfois être remplacé par un autre sans que le comportement de la protéine soit considérablement modifié, mais cela aura cependant la conséquence qu’un autre acide aminé du secteur pourra faire évoluer le comportement allostérique vers un comportement complètement différent lors d’une mutation ultérieure. Cette information « latente » explique pourquoi le phénotype bande passante/bande d’arrêt a révélé que ce sont des mutations covariantes qui engendrent une architecture interne coopérative au sein de la structure de la protéine. Les acides aminés correspondants forment des réseaux spécifiques d’acides aminés dans la structure de la protéine et influencent à distance la fonction du site actif. Ce comportement est cohérent avec une architecture des secteurs constituant une sorte de fil conducteur reliant les sites importants le long d’un chemin tridimensionnel constitué de certains acides aminés éloignés dans le squelette de la protéine plutôt que contigus dans le polypeptide [16]. À ce point de l’interprétation des données, il devient intéressant de noter que les secteurs impliqués dans les transitions allostériques ne se trouvent pas seulement dans les protéines, ils caractérisent aussi les molécules d’ARN fonctionnelles [17]. Cette nouvelle observation laisse entrevoir une raison d’être critique dans ce processus : il faut penser qu’une propriété générale, associée à des polymères biologiques très différents, protéines et acides nucléiques, soit apte à lier leurs propriétés fonctionnelles et leur structure. La question se pose alors de savoir ce qui pourrait être à l’origine de cette propriété omniprésente.
5. L’eau et les liaisons hydrogène comme interconnexion entre sites actifs
De nombreux processus pourraient expliquer la formation des secteurs. Une idée rarement explorée consisterait à faire intervenir l’eau. L’eau est le support universel de la vie, et c’est peut-être pour cette raison qu’elle est plus ou moins « invisible » dans les études structurales. La plupart des analyses par diffraction des rayons X présentent les protéines ou les acides nucléiques comme s’ils se trouvaient dans le vide, avec quelques molécules d’eau ici et là. Pourtant, le rôle de l’eau va bien au-delà de son rôle de solvant continu ou de son moment dipolaire. L’eau en contact avec une protéine a ses protons engagés dans un réseau de liaisons hydrogène (Figure 3). Cette caractéristique structurelle est bien visible dans la résonance des protons mesurée dans les études RMN [18]. Le réseau de protons qui entoure les macromolécules et engagés dans des liaisons hydrogène rappelle fortement les états possibles d’un « modèle d’Ising » (voir [19], comme exemple des nombreuses occurrences de la question en biologie). Dans le modèle d’Ising, chaque site du réseau n’interagit qu’avec les sites qui lui sont directement adjacents, tout en étant affecté par un champ général dans lequel l’objet de l’étude est intégré.
Illustration de la présence de molécules d’eau identifiées dans un cristal de protéine. La protéine-tyrosine phosphatase 1B d’Homo sapiens a été cristallisée à une résolution de 1,95 Å (entrée PDB 1SUG, https://www.rcsb.org/3d-view/1SUG/). Le squelette polypeptidique est illustré et les molécules d’eau qui ont pu être identifiées dans le cristal sont représentées par des points rouges. On peut voir sur la figure que la distribution de ces molécules n’est pas aléatoire et suit le repliement de la protéine.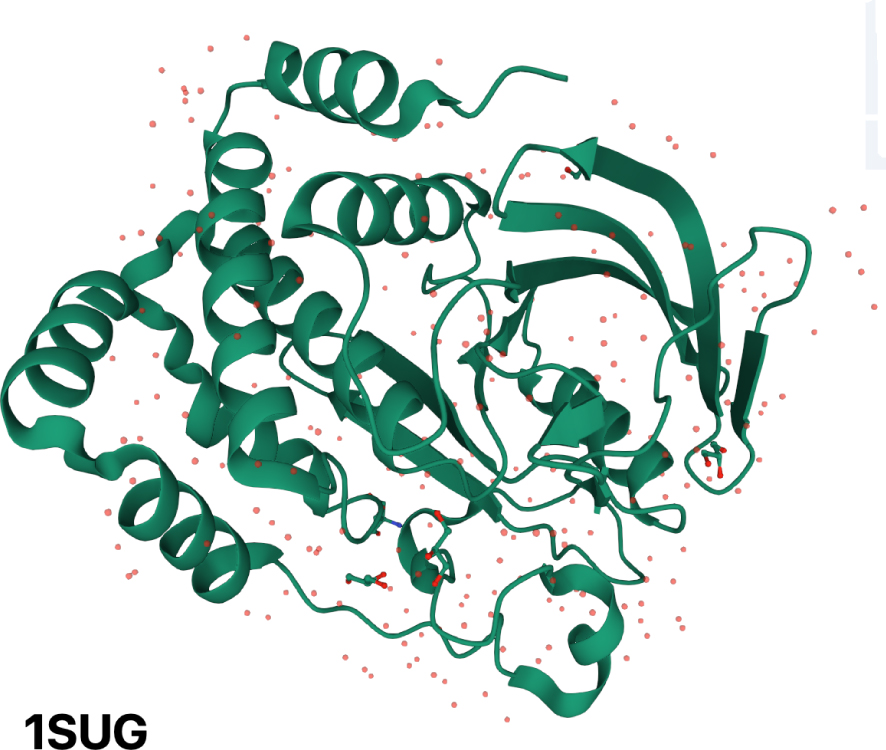
À 300 K et dans un environnement cellulaire particulier, il existe un équilibre entre les états possibles des liaisons hydrogène contraints par une macromolécule, et les états dominants sont généralement fortement déplacés dans le sens de la stabilisation d’une structure 3D particulière. Maintenant, les liaisons hydrogène peuvent passer d’un accepteur à l’autre très rapidement (l’énergie impliquée est de l’ordre de l’énergie thermique). Cela peut se produire de manière concertée, sous l’effet d’interactions avec des acides aminés spécifiques du biopolymère, mais aussi avec le squelette polypeptidique, passant d’une structure 3D à une autre (typiquement la situation MWC décrite en 1965), en fonction de la présence de substrats ad hoc dans l’environnement. Au cours de la transition, la formation et la rupture de liaisons hydrogène doivent parcourir une séquence spatio-temporelle, contraignant localement la chaîne des acides aminés de la protéine concernée. Cet effet peut se produire rapidement, mais parce que le déplacement des liaisons hydrogène doit se développer séquentiellement suivant une sorte d’« effet domino » [20], il doit opérer dans un laps de temps considérablement plus lent que les vibrations des atomes constituant la protéine (le taux d’échange d’une molécule d’eau prend des millisecondes ou même des secondes, et le basculement d’un proton d’une liaison hydrogène à une autre sera du même ordre de grandeur). En termes physico-chimiques et de façon analogue à la transition qui fait passer d’un état d’Ising à un autre état d’Ising, ce basculement peut être couplé à un changement mécanique illustré par une transition à travers des régions flexibles de la protéine, avec finalement rééquilibrage des liaisons hydrogène à plus grande distance, principalement au sein de l’eau, loin du squelette et des acides aminés de la protéine.
Dans ce contexte, nous pouvons proposer ici que le rôle des secteurs est de maintenir une organisation locale des molécules d’eau de telle sorte qu’elles forment un réseau de liaisons hydrogène, à la fois entre les molécules d’eau et la protéine et entre les molécules d’eau. Cela correspond aux catégories de mutants de LacI et à leurs transitions. Les auteurs de l’étude illustrée plus haut n’ont pas proposé d’explication physico-chimique à leur observation, mais elle est très probablement associée à l’interaction de l’eau avec la protéine, dans un scénario semblable aux « fils » de protons identifiés dans le complexe respiratoire [21]. L’originalité des filtres de type « U » peut alors être expliquée par un changement séquentiel d’un réseau de liaisons hydrogène qui s’inverse ou bifurque vers une autre structure lorsque la concentration de l’effecteur augmente.
Les auteurs de l’étude LacI ont noté que ce comportement est lié à un certain « désordre » dans la séquence locale de la protéine, une caractéristique qui est directement dérivée de la structure locale de l’eau. Nous venons de discuter de la façon dont les transitions allostériques nécessitaient la présence d’un état transitoire instable d’une partie de la protéine allostérique en cause. La nature de ce désordre — qu’il serait préférable de nommer « flexibilité » plutôt que désordre, car le concept même d’ordre implique une information cachée fournie par l’observateur — a clairement quelque chose à voir avec la réorganisation locale de l’eau. Ce type de transition ouvre temporairement la possibilité pour les molécules d’eau de former des liaisons hydrogène transitoires avec le squelette du polypeptide ou des chaînes latérales d’acides aminés spécifiques. Cela a pour conséquence que la dynamique des changements entropiques liés à la structure de l’eau peut contribuer de manière importante à expliquer un phénotype comme celui des filtres passe-bande ou coupe-bande. Lorsqu’un ligand atteint un site de liaison dans la protéine, il s’échange contre quelques molécules d’eau, ce qui déclenche une chaîne de rupture et de formation de liaisons hydrogène qui déclenchera une transition de l’état initial de la protéine vers un autre état. On notera que cette transition, déclenchée par les fluctuations thermiques, peut aussi exister en l’absence de ligand, éventuellement fortement déplacée dans une direction plutôt qu’une autre. Le rôle du ligand sera alors de sélectionner un état particulier, de sorte que l’équilibre est déplacé jusqu’à ce que tous les sites de liaison possibles soient occupés par le ligand (c’est ce qui est décrit dans le modèle MWC et discuté au début de ce texte). Si l’on accepte cette façon de voir, il n’y a nulle « instruction » fournie par le ligand pour dire à la protéine de changer de forme, mais sélection d’une forme éventuellement mineure, suivie d’un rééquilibrage du mélange.
6. Un autre regard sur l’eau : support d’information permettant la mémorisation et le rappel, l’eau ne doit avoir qu’une mémoire très courte
Une dernière remarque. Pour des raisons sociologiques qu’il conviendrait d’explorer, l’eau, élément si répandu qu’on en oublie les propriétés très originales — notons que son volume augmente lorsqu’elle gèle — a fait l’objet d’un déluge de pensées magiques, notamment chez les personnes peu intéressées par la connaissance scientifique. L’idée farfelue que ce solvant pourrait conserver la mémoire à long terme d’événements passés a été un joli prétexte pour les Britanniques pour se moquer de la crédulité des médias français [22]. Pourtant, il existe bel et bien un lien entre l’eau et la mémoire, mais son rôle est exactement à l’opposé de celui d’une mémoire à long terme, puisque la mémoire de l’eau est sujette à un effacement rapide. L’eau agit comme support permettant de véhiculer et d’effacer très rapidement le souvenir de ses états, comme vecteur général des réseaux de liaisons hydrogène qui sont utilisés pour véhiculer de façon invisible l’information biologique. Vue ainsi, l’eau permet aux cellules de calculer, comme le font les électrons dans les appareils électroniques. Avec cette remarque nous souhaitons mettre en avant le rôle crucial de l’eau dans les études biologiques, faisant que les analyses qui représentent les objets biologiques comme s’ils étaient dans un espace vide sont condamnées à passer à côté d’une des propriétés les plus importantes de la vie.