

1 Introduction
The suborder of snakes (Serpentes) includes about 3000 extant species 〚1〛. This richness is associated with a severely constrained bauplan that has always hampered morphological studies. At the same time, it has made available several modes of locomotion, diet and ecological niches. Nevertheless, after more than 100 years of research, several higher-level phylogenetic relationships appear to be established (Fig. 1) 〚2–6〛. Snakes are then divided into two major clades: the Scolecophidia and the Alethinophidia. The Scolecophidia (families Leptotyphlopidae, Typhlopidae, Anomalepididae) are small fossorial snakes with a limited gape size, which feed mainly on ants and termites 〚4〛. The remaining snakes are the Alethinophidia, which are the ‘typical’ snakes. They are characterised by their independent mandibles and their general ability to ingest prey of relative large size 〚7〛. The Alethinophidia include the fossorial ‘Anilioidea’ (Uropeltidae, Anilius, Anomochilus, Cylindrophis), whose monophyly is not ascertained and the Macrostomata, which include Xenopeltis, Loxocemus, Xenophidion, several lineages of ‘booids’ (Erycinae, Boinae, Pythoninae, Tropidophiinae, Ungaliophiinae, Bolyeriidae) and the Caenophidia. The most distinctive evolutionary trend within the Alethinophidia is the increase of the gape size 〚7〛.
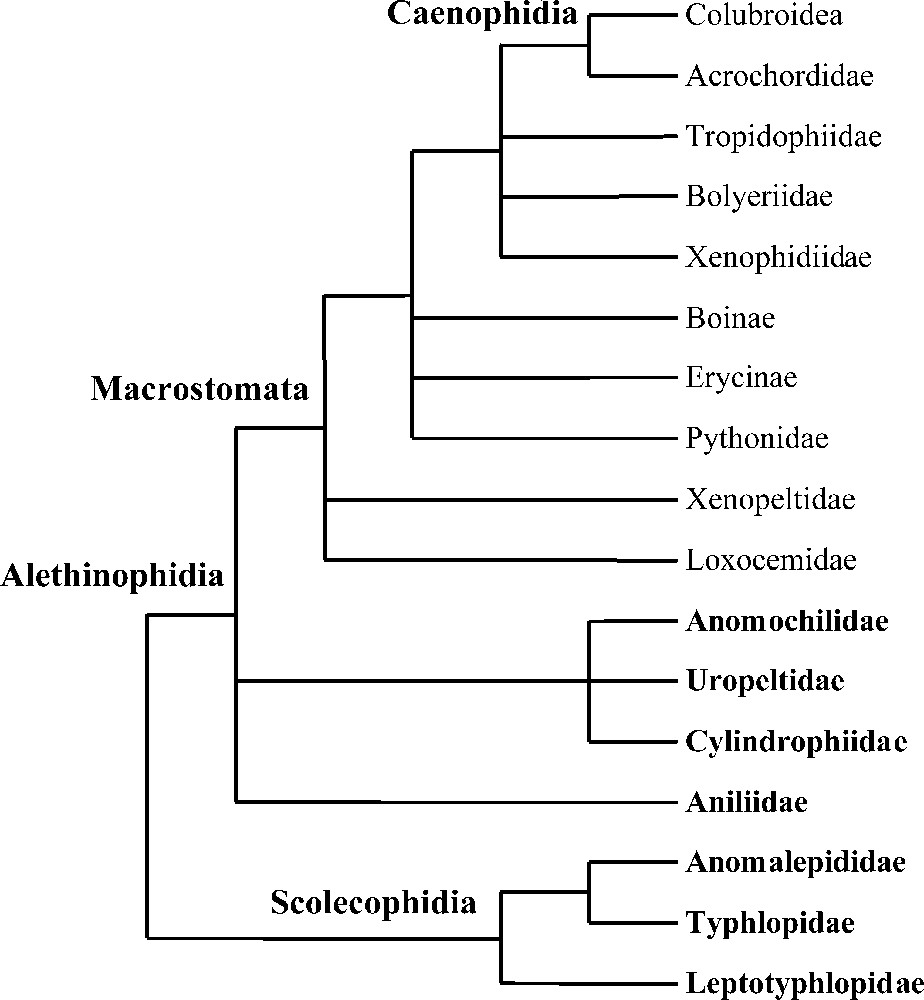
Phylogenetic relationships of snakes based on Cadle et al. 〚2〛, Cundall et al. 〚3〛, Wallach 〚4〛, Scanlon and Lee 〚5〛 and Tchernov et al. 〚6〛. Terminal taxa written in bold are fossorial (non-macrostomatan).
In spite of these advances, several multifurcations remain: interrelationships of ‘anilioid’ lineages and interrelationships of most macrostomatan lineages. The aim of this work is first to shed light on these unresolved points of snake phylogeny using DNA sequences and then to address the following evolutionary question: what was the ancestral mode of life of snakes? For this purpose, 136 sequences (85 original) were used, obtained from one nuclear and three mitochondrial genes and representing all major snake lineages but two: Anomochilidae (genus Anomochilus) and Xenophidiidae (genus Xenophidion), both being known only from a handful of specimens.
2 Materials and methods
2.1 DNA extraction, PCR and sequencing
Tissue samples (tissue homogenate, liver, blood, tail tip, or shed skin) were obtained from the tissue collections of Nicolas Vidal and S. Blair Hedges (see Appendix 1). DNA extraction followed protocols previously described 〚8〛. Amplification was performed using the following sets of primers: L2510, 5’–CGC–CTG–TTT–ATC–AAA–AAC–AT–3’ 〚9〛, L16, 5’–ACG–GCC–GCG–GTA–YCC–TAA–CCG–TG–3’ (original) and H3056, 5’–CTC–CGG–TCT–GAA–CTC–AGA–TCA–CGT–AGG–3’ 〚10〛 for the 16S rRNA gene; L12, 5’–CGC–CAA–AYA–ACT–ACG–AG–3’ (original), H1478, 5’–TGA–CTG–CAG–AGG–GTG–ACG–GGC–GGT–GTG–T–3’ 〚11〛 and H1557, 5’–GTA–CAC–TTA–CCT–TGT–TAC–GAC–TT–3’ 〚12〛 for the 12S rRNA gene; L39, 5’–CTG–SAR–YTT–TCT–YCA–TCT–GT–3’ (original), HC3, 5’–CAA–ACA–TTA–YRT–TCT–GTG–ATG–A–3’(original) and G74, 5’–TGA–GCA–TCC–AAA–GTC–TCC–AAT–3’ 〚13〛 for the C-mos gene; L14724, 5’–TGA–CTT–GAA–GAA–CCA–CCG–TTG–3’ 〚9〛, LLIO, 5’–AAC–ATC–TCA–RCM–TGA–TGA–AA–3’ (original) and HVN650, 5’–TAT–GGG–TGG–AAK–GGG–ATT–TT–3’ (original) for the cytochrome b gene. Both strands of the PCR products were sequenced using the CEQ cycle sequencing kit (Beckman) in the CEQ-2000 DNA Analysis System (Beckman). The two strands obtained for each sequence were aligned using the BioEdit Sequence Alignment Editor program 〚14〛. Sequence data obtained from Genbank or bibliography are listed in Appendix 2.
2.2 Sequence analysis
Sequence entry and alignment were performed manually with the MUST2000 software 〚15〛. After removal of the 5' end of the cytochrome b gene, alignment was straightforward as there were no indels. For the C-mos gene, amino acid properties were used, resulting in an alignment including one codon deletion defining Serpentes, one codon deletion defining Typhlopidae and one deletion defining Alethinophidia (one to three codons in length according to alethinophidian taxa). For the 16S rRNA sequences, alignment was ambiguous in three highly variable areas, corresponding to loops that we have deleted from analyses. In order to align the 12S rRNA sequences, we used the secondary structure model described by Hickson et al. 〚16〛. The alignments will be deposited in EMBL alignment database and the complete sequences will be deposited in GenBank upon publication. In all further analyses, gaps were excluded. We followed the approach outlined by Huelsenbeck and Crandall 〚17〛 to test alternative models of evolution, using PAUP* 〚18〛 and Modeltest 〚19〛. A starting tree was obtained by NJ 〚20〛. With this tree, likelihood scores were calculated for 56 models of evolution and then compared statistically using a chi-square test with degrees of freedom equal to the difference in free parameters between the models being tested. Once a model of evolution was chosen, it was used to estimate a tree using the minimum evolution optimality criteria 〚21〛. Support for nodes was then estimated using the bootstrap technique 〚22〛, with 2000 replicates. All phylogenetic analyses were performed with PAUP*. The separate analyses showed no significant topological incongruence (no conflicting nodes exhibited a bootstrap value above 70%). We performed combined analyses after having defined corresponding models of evolution using the procedure described above. Results from the latter are presented under the form of bootstrap consensus trees (2000 replicates), which are considered as reliable estimates of phylogeny 〚23〛. Bootstrap values measure internal robustness only; the accuracy of nodes was estimated using taxonomic congruence between independent datasets (such as nuclear and mitochondrial markers or molecular and morphological data).
3 Results and discussion
3.1 Models of evolution selected
The C-mos data set includes 575 bp for 76 taxa (333 variable sites, 246 of which are informative for parsimony) covering the diversity of snake lineages (including caenophidian ones). The model selected is the HKY (Hasegawa, Kishino and Yano 〚24〛) +I+G model (base frequencies: A (0.278), C (0.207), G (0.223), T (0.292); TS/TV ratio: 2.55; proportion of invariable sites (I): 0.32; gamma distribution shape parameter (G): 2.5). The 12S–16S rRNA dataset includes 679 bp for 70 taxa (379 variable sites, 327 of which are informative for parsimony) covering the diversity of snake lineages (including caenophidian ones). The model selected is the GTR (General Time Reversible 〚25〛) +I+G model (base frequencies: A (0.427), C (0.212), G (0.17), T (0.192); rate matrix: 〚AC〛: 16.21, 〚AG〛: 25.11, 〚AT〛: 8.02, 〚CG〛: 1.93, 〚CT〛: 92,25, 〚GT〛: 1; I: 0.38; G: 0.64). The cytochrome b data set includes 574 bp for 31 taxa (369 variable sites, 327 of which are informative for parsimony) covering the diversity of snake lineages. The selected model is the TVM (Transversional 〚25〛) +G model (base frequencies: A (0.396), C (0.365), G (0.059), T (0.181); rate matrix: 〚AC〛: 0.09, 〚AG〛: 4.66, 〚AT〛: 0.36, 〚CG〛: 0.35, 〚CT〛: 4.66, 〚GT〛: 1; G: 0.18). The combined C-mos/12S/16S rRNA data set includes 1257 bp for 66 taxa (710 variable sites, 566 of which are informative for parsimony) covering the diversity of snake lineages (including caenophidian ones). The model selected is the GTR+I+G model (base frequencies: A (0.349), C (0.209), G (0.199), T (0.243); rate matrix: 〚AC〛: 4.88, 〚AG〛: 7.88, 〚AT〛: 2.36, 〚CG〛: 1.31, 〚CT〛: 20.08, 〚GT〛: 1; I: 0.26; G: 0.48). The combined C-mos/12S–16S rRNA/cytochrome b dataset includes 1840 bp for 31 taxa (1027 variable sites, 773 of which are informative for parsimony) covering the diversity of snake lineages. The model selected is the GTR+I+G model (base frequencies: A (0.341), C (0.286), G (0.158), T (0.215); rate matrix: 〚AC〛: 2.81, 〚AG〛: 5.79, 〚AT〛: 2.11, 〚CG〛: 0.62, 〚CT〛: 15.11, 〚GT〛: 1; I: 0.21; G: 0.41). It should be noted that the model selected for the C-mos (protein coding nuclear gene with even base composition) analysis is simpler than the one selected for the cytochrome b (protein coding mitochondrial gene with uneven base composition) analysis, which is itself simpler than the models selected for the 12S–16S rRNA (mitochondrial genes encoding ribosomal RNA with uneven base composition and complex secondary structure) and combined gene analyses.
3.2 Phylogenetic results
For reason of space limitations, the bootstrap consensus ME tree (2000 replicates) obtained from the combined analysis including four genes is presented only (C-mos/12S–16S rRNA/cytochrome b). All results discussed below are based on this tree except otherwise mentioned.
3.2.1 Higher-level snake relationships
Scolecophidia form a monophyletic group with Leptotyphlopidae (Leptotyphlops) as sister-group to a clade formed by Anomalepididae (Liotyphlops) and Typhlopidae (Ramphotyphlops) (Fig. 2). Although not strongly supported by bootstrap values, we consider this topology to be reliable as it is identical to the one obtained by Wallach 〚4〛 from an extensive anatomical work. The monophyly of Alethinophidia 〚26〛 is strongly supported by our molecular dataset (bootstrap value: 100%). The early divergence of snakes into two lineages very distinct morphoecologically (fossorial Scolecophidia feeding on small prey on a frequent basis versus ecologically diverse Alethinophidia feeding on large prey on an infrequent basis) is then confirmed by our data. Within Alethinophidia, the most surprising result from our study is the clustering of the Tropidophiidae sensu stricto (genera Tropidophis and Trachyboa (sister-groups based on 12S–16S rRNA analysis (bootstrap value: 100%) with the Aniliidae (genus Anilius) (bootstrap value: 98%). Indeed, Tropidophis and Trachyboa are considered to belong to the Macrostomata, a clade strongly supported by morphology, which includes the representatives of Alethinophidia that are not ‘anilioids’ (Anilius, Cylindrophis, Rhinophis and Uropeltis in our dataset) 〚5, 6, 27〛. Our molecular result then disagrees with morphological evidence (see Fig. 1). Nevertheless it is biogeographically coherent (Tropidophis, Trachyboa and Anilius are all from the Neotropics) and is supported by all genes analysed separately. Moreover, it is not the result of a contamination event, as it was independently found by Campbell in his PhD study using cytochrome b sequences obtained from tissue samples different from ours 〚28〛. The consequence of this result concerning the early evolution of snakes is discussed below. In any case, ‘anilioids’ as presently defined include two lineages, an American one (Anilius, Tropidophis, Trachyboa) and an Asiatic one (Cylindrophis, Uropeltidae represented here by Rhinophis and Uropeltis, and Anomochilidae – genus Anomochilus, which is not included in our taxonomic sample but is conservatively considered to belong to this clade according to morphology (see Fig. 1) and distribution). The next higher ranked clade includes two main groups: the Caenophidia (bootstrap value: 66%) and a clade formed by various lineages of ‘booids’. Even if this last node is not strongly supported by our data (bootstrap value below 50%), the grouping of pythons, boas, and associated lineages (Bolyeriidae, Loxocemidae, Xenopeltidae) had already been favoured by some morphologists 〚27, 29–33〛. Moreover, those two clades correspond to two distinct large-size prey neutralisation modes: unimodal constriction for ‘booids’ (locomotor and feeding systems coupled) and injection of toxic saliva, in addition (or not) to diverse alternate modes of constriction, for Caenophidia (locomotor and feeding systems uncoupled; see our companion paper on caenophidian relationships) 〚34〛. We therefore use the term Henophidia (first introduced by Hoffstetter 〚29〛 to include Alethinophidia not belonging to Caenophidia) to describe the clade including ‘booids’. It should be noted that the C-mos alone analysis gives an alternative topology with the henophidian clade including not only ‘booids’, but also ‘anilioids’ as presently defined (which then appear in a basal position among Henophidia, and which use the same mode of constriction as ‘booids’ when they constrict), i.e. all non-caenophidian Alethinophidia (bootstrap value below 50%). Within the Henophidia, the Bolyeriidae (characterised by an intramaxillary joint, a unique feature among vertebrates 〚35〛 and represented here by Casarea dussumieri, the sole extant species of the family, living at the Île Ronde, off the coast of Mauritius) are in a basal position. The monophyly of pythons (genera Python, Liasis, Apodora in combined analyses, plus Morelia sequenced for 12S–16S rRNA and C-mos, but not for cytochrome b) is strongly supported (bootstrap value: 100%). The closest relative of pythons consists of a clade of Xenopeltis and Loxocemus (bootstrap values: 65%), a result that had already been proposed by some morphologists 〚32, 33〛. The position of Calabaria as closest relative of the boas, moderately supported by our data (bootstrap value: 59%), contradicts the hypothesis that Calabaria belongs to erycids (a family of boas) 〚36〛. Within boas, the boids appear to be paraphyletic (genera Boa, Acrantophis, Candoia), while the erycids are monophyletic (genera Eryx, Exiliboa, Gongylophis, Ungaliophis, Lichanura and Charina). Ungaliophis and Exiliboa (from the Neotropical area) cluster with the North American erycid genera (Charina and Lichanura, bootstrap value: 90%) while the two Old World representatives form another clade (genera Eryx and Gongylophis, bootstrap value: 99%). Although many herpetologists consider Exiliboa and Ungaliophis as representatives of the tropidophiids (with the Neotropical genera Tropidophis and Trachyboa), Zaher 〚37〛 had already shown on a morphological basis that the tropidophiids were not monophyletic, but consisted of two very distinct lineages: Trachyboa and Tropidophis, on the one hand, (tropidophiids) and Ungaliophis and Exiliboa, on the other hand (‘booids’).
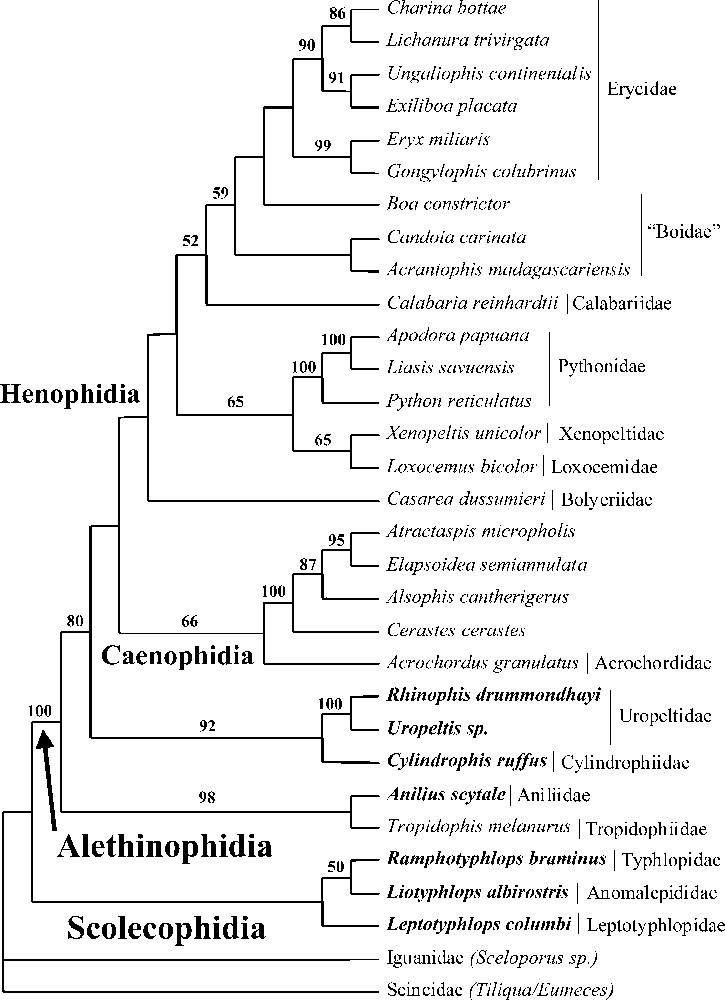
Phylogenetic relationships of snakes based on C-mos, 12-16S rRNA and cytochrome b sequences (bootstrap ME consensus tree, 2000 replicates, values above 50% are shown). Terminal taxa written in bold are fossorial (non macrostomatan). The C-mos alone analysis gives an alternative topology where the two ‘anilioid’ lineages are not basal alethinophidian snakes, but basal henophidian snakes.
3.2.2 Evolutionary implications: the ancestral mode of life of snakes
Three main hypotheses have been proposed concerning the ancestral mode of life of snakes: fossorial, terrestrial, or marine 〚38–41〛. This controversy, which has profound implications on our understanding of the evolution of locomotor and feeding systems in snakes, has recently been fueled by the discovery of several marine fossils with well-developed hindlimbs 〚6, 42, 43〛. The phylogenetic position of these fossils is hotly debated as they could either be the closest relative of snakes (according to Scanlon and Lee 〚5〛, Caldwell and Lee 〚42〛, Rage and Escuillié 〚43〛 and Rage 〚44〛, favouring a marine origin of snakes) or derived macrostomatan snakes (according to Tchernov et al. 〚6〛 and Zaher and Rieppel 〚45–47〛, favouring a terrestrial/fossorial origin). Although solving the position of snakes among squamates and testing the marine origin hypothesis are out of the scope of this work, mapping extant modes of life (fossorial i.e. ability to burrow into the soil versus terrestrial) on the phylogeny depicted in Fig. 1 reveals a very significant fact. All non-macrostomatan snakes (microstomatan scolecophidians and traditional ‘anilioids’) are fossorial, a mode of life associated with a compact skull limiting the choice of ingestible prey (small and/or elongated) while the phylogenetically derived macrostomatan clade is of terrestrial origin. The distribution of the fossorial/terrestrial traits then corresponds exactly to the non-macrostomatan/macrostomatan categories. The classical evolutionary trend within the Alethinophidia, the increase in gape size (which results from one fossorial to terrestrial transition) was the result of excluding fossorial alethinophidian lineages from the definition of Macrostomata. Based on our phylogeny, whether the ancestral snake was fossorial or terrestrial requires two or three evolutionary events respectively: two fossorial to terrestrial transitions (fossorial ancestor) or three terrestrial to fossorial transitions (terrestrial ancestor) (Fig. 2). A fossorial origin of snakes then appears to be the most parsimonious hypothesis. Nonetheless, this option is only one step more parsimonious than the alternative one and implies the independent acquisition of the very complex macrostomatan condition in two lineages. Therefore, the hypothesis of a non-fossorial/macrostomatan origin of snakes with three subsequent transitions to a fossorial/non-macrostomatan condition (a common event among vertebrates in general and squamates in particular 〚44, 48〛) should remain a viable alternative for future consideration. Moreover, fossorial ‘anilioids’ would not represent a kind of intermediate stage between ‘true’ microstomatan Scolecophidia and ‘true’ macrostomatan Alethinophidia but would be regressed macrostomatan snakes, a hypothesis also supported by the C-mos alone analysis (where the two ‘anilioid’ lineages are basal henophidian snakes). Although our data are insufficient to robustly infer the ancestral mode of life of snakes, we find evidence of plasticity in the basic ecological and trophic modes of snakes. Consequently, the macrostomatan condition should not be treated a priori as a derived character state devoid of homoplasy.
Acknowledgements
This work was supported for its most part by the ‘Service de systématique moléculaire’, ‘Institut de systématique’ (CNRS FR 1541), directed by Simon Tillier. N.V. thanks C. Bonillo, K. Daoues, P. David, R. Debruyne, A. Dubois, A. Halimi, G. Lecointre, O. Pauwels, F. Pleijel, J.-C. Rage, A. Tillier, S. Tillier for their help during the course of this work. S.B.H. thanks D. Rabosky for technical assistance and NSF and NASA for partial support. N.V. and S.B.H. thank the following persons for contributing most of the tissue samples used in this work: M. Boulay, R. M. Burger, B. N. Campbell, L. Chirio, K. Daoues, I. Das, P. David, S. Delahaie Thourin, J.-C. de Massary, H. G. Dowling, C. Gans, A. Halimi, S. Imbott, I. Ineich, U. Kuch, P. Lacour, S. Lavoué, O. Le Duc, D. Mebs, T. Moncuit, P. Moret, O. Pauwels, J. Reynes, C. Skliris, A. Teynié, W. Wüster, H. Zaher.
Version abrégée
Le sous-ordre des serpents (Serpentes) comprend environ 3000 espèces actuelles, occupant des niches écologiques variées. Cette richesse, associée à un plan de base très contraint, a toujours rendu l’étude morpho-anatomique des serpents délicate. Cependant, après plus de cent ans de recherche, plusieurs relations phylogénétiques de taxons de haut rang taxinomique semblent établies. Ainsi les serpents sont divisés en deux grands clades : les Scolecophidia et les Alethinophidia. Les Scolecophidia (familles des Leptotyphlopidae, Typhlopidae et Anomalepididae) sont des serpents fouisseurs de petite taille, présentant une ouverture de la bouche limitée et se nourrissant principalement de termites et de fourmis. Les Alethinophidia sont les serpents « typiques », qui sont caractérisés par l’indépendance de leurs mandibules et leur capacité à ingérer des proies plus grosses que le diamètre de leur propre corps. Les Alethinophidia comprennent les « Anilioidea » (Uropeltidae, Anilius, Anomochilus, Cylindrophis), tous fouisseurs, dont la monophylie n’est pas établie de façon fiable et le clade des Macrostomata (« serpents à grande bouche »), qui comprend Xenopeltis, Loxocemus, Xenophidion, plusieurs lignées de « Booidea » (Erycinae, Boinae, Pythoninae, Tropidophiinae, Ungaliophiinae, Bolyeriidae) et les Caenophidia (qui incluent tous les serpents venimeux). Ainsi, la tendance évolutive la plus remarquable au sein des Alethinophidia est l’augmentation progressive de l’ouverture de la bouche, depuis les « Anilioidea » jusqu’aux Caenophidia.
Malgré ces avancées dans notre connaissance, de nombreuses relations de parenté demeurent irrésolues, en particulier entre les lignées d’« Anilioidea » et entre les lignées de Macrostomata. Le but de ce travail consiste d’abord à éclairer ces points en utilisant des séquences d’ADN, puis à déterminer quel était le mode de vie ancestral des serpents. Pour cela, 136 séquences (dont 85 d’entre elles sont originales) ont été utilisées, obtenues à partir d’un gène nucléaire (C-mos) et de trois gènes mitochondriaux (ARNr 12S et 16S, cytochrome b), et représentant toutes les lignées actuelles de serpents, sauf deux d’entre elles, qui ne sont connues que par un très faible nombre de spécimens : les Anomochilidae (genre Anomochilus) et les Xenophidiidae (genre Xenophidion).
Cinquante-six modèles alternatifs d’évolution moléculaire ont d’abord été testés de façon statistique pour chacun des gènes utilisés à l’aide d’une approche de maximum de vraisemblance. Le modèle choisi a alors été utilisé pour l’estimation des phylogénies, en utilisant le critère d’optimalité du minimum d’évolution. La robustesse des nœuds a été estimée à l’aide de la technique du bootstrap, avec 2000 réplicats. Les analyses séparées ne montrant pas de non-congruence topologique significative (aucun nœud contradictoire soutenu par des valeurs de bootstrap supérieures à 70%), nous avons réalisé des analyses combinées en répétant la procédure décrite ci-dessus. La technique de bootstrap ne mesurant que la robustesse interne d’un jeu de données, la fiabilité des nœuds a été estimée en utilisant le critère de congruence taxinomique entre jeux de données indépendants.
Les Scolecophidia forment un groupe monophylétique avec les Leptotyphlopidae, groupe frère des Anomalepididae et des Typhlopidae. Les Alethinophidia sont également retrouvés monophylétiques. La divergence précoce des serpents en deux lignées très différentes sur le plan morpho-écologique (Scolecophidia fouisseurs, se nourrissant de petites proies, avec une fréquence rapprochée des repas, et Alethinophidia écologiquement variés, se nourrissant de grosses proies, avec une fréquence espacée des repas) est donc confirmée par nos données.
Au sein des Alethinophidia, le résultat le plus surprenant de notre étude est la relation de groupes frères des Tropidophiidae (genres Tropidophis et Trachyboa) et des Aniliidae (genre Anilius). En effet, selon les études morphologiques, les Tropidophiidae appartiennent au clade des Macrostomata, qui comprend les Alethinophidia non « Anilioidea » (Uropeltidae, Anilius, Anomochilus, Cylindrophis). Ce résultat est donc en désaccord avec les données morphologiques, mais est cohérent d’un point de vue biogéographique (les Tropidophiidae et les Aniliidae sont tous néotropicaux). Ainsi, les « Anilioidea » sont constitués de deux lignées distinctes : une lignée américaine (néotropicale) (genres Anilius, Trachyboa et Tropidophis) et une lignée asiatique (Uropeltidae, genres Anomochilus et Cylindrophis).
Les autres représentants des Alethinophidia se répartissent en deux grands clades : le clade des Caenophidia et un clade regroupant les différentes lignées de « Booidea », que Robert Hoffstetter avait pour la première fois reconnu sous le nom d’Henophidia. Ces deux clades correspondent à deux modes distincts de neutralisation des proies : constriction unimodale chez les Henophidia (structures de nutrition et de locomotion couplées) et injection de salive toxique, associée ou non à divers modes de constriction alternatifs, chez les Caenophidia (structures de nutrition et de locomotion découplées). Il est à noter que l’analyse du gène nucléaire C-mos aboutit à une topologie différente de celle issue de l’analyse combinée des quatre gènes, puisque, dans ce cas, le clade des Henophidia n’inclut pas seulement les lignées de « Booidea », mais aussi les lignées d’« Anilioidea » (qui apparaissent alors en position basale au sein des Henophidia et qui utilisent, lorsqu’elles constrictent, le même mode de constriction que les « Booidea »), c’est-à-dire toutes les lignées d’Alethinophidia à l’exception des Caenophidia.
Au sein des Henophidia, les Bolyeriidae (caractérisés par une articulation intra-maxillaire et représentés par Casarea dussumieri, la seule espèce actuelle de la famille, endémique de l’île Ronde au large de l’île Maurice) occupent une position basale. Les pythons forment un groupe monophylétique dont le groupe frère est constitué par le clade comprenant les genres Loxocemus et Xenopeltis. L’énigmatique genre africain Calabaria est groupe frère des boas, qui forment un groupe monophylétique. Au sein des boas, les Boidae apparaissent paraphylétiques. Les Erycidae sont monophylétiques et comprennent les genres néotropicaux Exiliboa et Ungaliophis, qui sont groupes frères des Erycidae nord-américains.
Les relations phylogénétiques au sein des Caenophidia font l’objet d’un article distinct. Ils apparaissent comme le groupe frère des Henophidia, rompant avec une conception des Henophidia groupe souche des Caenophidia.
Concernant le mode de vie ancestral des serpents, trois hypothèses principales ont été avancées : une origine fouisseuse, une origine terrestre et une origine marine. Bien qu’il nous soit impossible de tester l’hypothèse de l’origine marine (qui repose entièrement sur des serpents fossiles), nos donnés nous permettent de remarquer que la distribution des modes de vie terrestre et fouisseur correspond exactement à la distribution des conditions macrostomate (grande bouche) et non-macrostomate (petite bouche). D’après l’arbre issu de l’analyse combinée des quatre gènes, que le mode de vie ancestral soit fouisseur ou non, deux ou trois événements, respectivement, sont requis : deux transitions de la vie fouisseuse à la vie terrestre dans le premier cas et trois transitions de la vie terrestre à la vie fouisseuse dans le second. Une origine fouisseuse des serpents apparaît donc comme l’hypothèse la plus parcimonieuse. Cependant, elle requiert l’acquisition indépendante de la condition macrostomate, très complexe, dans deux lignées. C’est pourquoi l’hypothèse d’une origine non fouisseuse des serpents, suivie par trois transitions vers le mode de vie fouisseur, un événement commun chez les vertébrés en général et les squamates en particulier, est à envisager. L’absence de la condition macrostomate chez certains « Anilioidea » résulterait de pertes secondaires liées à l’acquisition du mode de vie fouisseur, une hypothèse également favorisée par l’arbre issu de l’analyse du gène nucléaire C-mos (dans lequel les « Anilioidea » occupent une position basale au sein des Henophidia). Dans tous les cas, la condition macrostomate ne devrait pas être considérée a priori comme dérivée et dépourvue d’homoplasie.
Appendix 1. Tissue samples used
Tissue samples were obtained from the tissue collection of Nicolas Vidal for the following species (sequences produced: C: C-mos, 12/16: 12/16S rRNA, CY: cytochrome b).
Acrantophis madagascariensis (Madagascar; C, 12/16), Acrochordus granulatus (〚MNHN 1997.6576〛, Ko Mai Phai Island, Muang District, Phang-Nga Province, Thailand; C, 12/16), Alsophis cantherigerus (Cuba; C, CY), Apodora papuana (Irian Jaya; C, 12/16), Atractaspis micropholis (Togo; C, 12/16), Boa constrictor (Petit Saut, French Guiana; C), Calabaria reinhardtii (Togo; C, CY), Candoia carinata (Halmahera Island, Indonesia; C, 12/16), Cerastes cerastes (captive born; C, 12/16), Charina bottae (captive born; C, 12/16), Cylindrophis ruffus (〚MNHN 1999.9021〛, Indonesia; C, 12/16, CY), Elapsoidea semiannulata (Central African Republic; C, 12/16), Eryx miliaris (unknown origin; C, 12/16), Gongylophis colubrinus (captive born; C, 12/16), Liasis savuensis (Savu Island, Indonesia; C, 12/16), Lichanura trivirgata (captive born; C, 12/16), Morelia boeleni (Wamena, Irian Jaya; C, 12/16), Python reticulatus (captive born, C), Ramphotyphlops braminus (〚NV RBR 001〛, Phang-Nga City, Muang District, Phang-Nga Province, Thailand; C, 12/16), Tiliqua scincoides (Indonesia; 16), Xenopeltis unicolor (〚CUB MZ R 1998.12.11.30〛, Ban Salakern, Ban Lat District, Phetchaburi Province, Thailand; C, 12/16, CY).
Tissue samples were obtained from the tissue collection of S. Blair Hedges for the following species:
Anilius scytale (SBH 267100, locality unknown; C, 12/16), Casarea dussumieri (SBH 267099, locality unknown; C, 12/16), Leptotyphlops columbi (SBH 192936, Little Fortune Hill, San Salvador, Bahamas; C, CY), Liotyphlops albirostris (SBH 172151, ‘Venezuela’; C, CY), Loxocemus bicolor (HGD 145976, ‘Mexico’; C, 12/16), Rhinophis drummondhayi (SBH 194102, north of Namunukula, Pindarawatta, Sri Lanka; C, CY), Trachyboa gularis (SBH 194899, locality unknown; 12/16), Tropidophis melanurus (SBH 172610, Soroa, Pinar del Rio, Cuba; C, 12/16), Typhlops jamaicensis (SBH 172445, 6.2 km west of Oracabessa, St. Mary, Jamaica; C, 12/16), Typhlops lumbricalis (SBH 191018, La Fangosa, Guantánamo, Cuba; 12/16), Ungaliophis continentalis (SBH 194642l, locality unknown; C, 12/16).
Appendix 2. Sequence data obtained from Genbank
Sequence data for the following genes and species were obtained from Genbank.
C-mos gene: Exiliboa placata (AY099973), Sceloporus grammicus (AF039478), Tiliqua scincoides (AF039462), Uropeltis phillipsi (AF471100).
12S and/or 16S rRNA gene: Alsophis cantherigerus (AF158475, AF158405), Boa constrictor (Z46470, Z46495), Calabaria reinhardtii (Z46464, Z46494), Exiliboa placata (AF512742), Leptotyphlops columbi (Z46488, Z46462), Liotyphlops albirostris (Z46487, Z46461), Python reticulatus (Z46448, Z46478), Rhinophis drummondhayi (Z46477, Z46447), Sceloporus grammicus (AF154130, L41464), Tiliqua scincoides (AF090187), Tropidophis wrighti (Z46445, Z46476), Uropeltis melanogaster (AF512739).
Cytochrome b gene: Acrantophis madagascariensis (U69736), Acrochordus granulatus (AF217841), Anilius scytale (U69738), Apodora papuana (U69843), Atractaspis micropholis (AF039261), Boa constrictor (U69740), Candoia carinata (U69753), Casarea dussumieri (U69755), Cerastes cerastes (AF039265), Charina bottae (U69757), Elapsoidea semiannulata (AF039260), Eryx miliaris (U69824), Eumeces egregius (AB016606), Exiliboa placata (AY099989), Gongylophis colubrinus (U69812), Liasis savuensis (U69839), Lichanura trivirgata (U69844), Loxocemus bicolor (U69845), Python reticulatus (U69859), Ramphotyphlops braminus (U69865), Sceloporus jarrovii (AF194219), Tropidophis melanurus (U69869), Ungaliophis continentalis (U69870), Uropeltis phillipsi (AF471034).