

1 Introduction
One of the central questions in biology is how genes act to form a complex organism from a single cell, the fertilized egg. Despite the enormous advances in our knowledge of molecular mechanisms of development that have taken place over the past two decades, human development is one of the most complex processes known, and the extent of what is understood is dwarfed by what remains unknown.
Mammalian development can be described as the progressive loss of totipotency followed by the loss of pluripotency, starting from the fertilized egg, with unlimited differentiation potential, to the differentiation of committed progenitor cells. This description reflects the fact that converting differentiated cells to pluripotent cells, a key problem for the future of stem cell-based therapy, is an ‘up-hill battle’ contrary to the usual mechanisms of cell differentiation. The only effective way to do this so far is Nuclear Transplantation, or animal cloning [1,2]. The concept of differentiation and epigenetic landscapes [3] is a useful way to organize what is known and speculated about the interactions between development, differentiation, and the genome, but molecular mechanisms are few and far-between in these landscapes, and major questions remain. For example, we know that differentiation potential varies from one cell population to the next, but what molecular determinants control or describe this? How are these mechanisms regulated?
The advent of genomics and bioinformatics raised the possibility of addressing these kinds of questions by looking at the actions of many genes simultaneously, rather than one gene at a time. To do this, we have been employing an ‘embryogenomics’ approach [4], a systematic analysis of genes expressed during development using large-scale genomics methodologies. The core of this approach is the Expressed Sequence Tag (EST) [5] project to produce complementary DNA (cDNA) libraries from embryonic tissues, combined with cDNA microarray analyses.
The large-scale human EST projects have been performed internationally and have accumulated more than 4 million ESTs [6–9]. The use of such resources was tremendously enhanced by the implementation of specialized public sequence databases (e.g., dbEST [10]) and the distribution of royalty-free cDNA clones to the community (e.g., IMAGE Consortium [11]). Large-scale mouse EST projects began much later in 1996 and have accumulated more than 2.5 million ESTs [12–14]. Within the EST collection field, our laboratory's particular emphasis is early mouse embryos, e.g., pre-implantation development and stem cells. It is particularly important to study this stage of development because it is not well represented in other databases for human or mouse transcripts (Fig. 1). For example, the earliest embryonic stage represented in public human EST databases as of 1 March 2002 was eight weeks post-ovulation. This stage corresponds to 14 days post-conception in mouse, at which point all of the critical developmental events such as pre-implantation, gastrulation, and organogenesis have already taken place, suggesting that genes with specific functions in these early stages are not likely to be included in human EST collections. This also means that there is a need for microarray platforms representing such genes, for the study of early embryos and stem cells. Ethical and technical issues make the use of human embryos at these early stages unfeasible; hence mouse is an important model organism for embryogenomic studies.
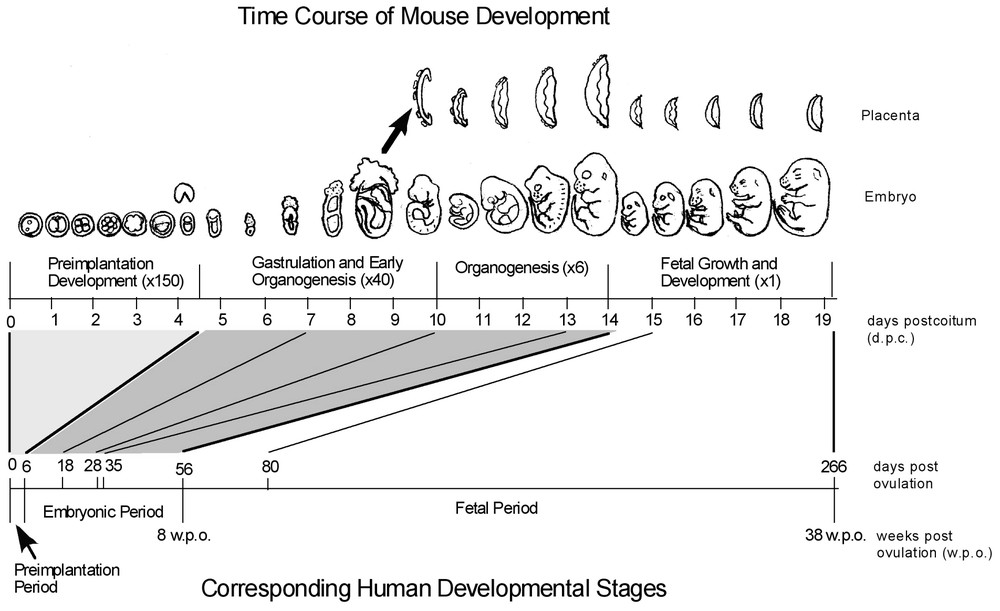
Comparison of mouse and human developmental time course. Mouse developmental progress is represented along the top axis in days post-coitus (dpc) and in representative images. Human development is marked in days post-ovulation on the bottom axis. Relative to the length of the entire gestation period, the critical periods of implantation, gastrulation, and organogenesis occupy a much greater share of developmental time in the mouse (two-thirds in mouse, compared to one-fifth in human). Combined with the fact that the earliest developmental time point represented in public human EST databases is eight weeks post-conception, it is clear that genes which have roles specific to early embryogenesis will not be isolated using human embryos. Mouse embryos, in contrast, allow for very effective isolation of early embryonic transcripts, including those expressed specifically in pre-implantation embryos. (Adapted from [4].)
In this review, we will first present a brief historical overview of our cDNA project, followed by a description of the resources and tools that have been developed as part of the project, as well as the current status of the project.
2 A brief historical account of the mouse cDNA project in our laboratory
We began to think about our mouse cDNA project in 1983, when one of the authors (M.S.H.K) realized the need for resources and technologies for global gene expression profiling to understand cell differentiation processes in molecular terms, particularly those involved in early mammalian development (Fig. 1). Two technical difficulties were anticipated at that time: (i) how to collect all the genes functioning in an organism without redundancy, and (ii) how to monitor the expression levels of individual genes with a membrane-based hybridization system. Because the latter had already been addressed for 100 genes in the pioneering work of Igor Dawid's group [15], the former problem seemed to be a greater challenge. Thus, we focused our efforts on how to collect all transcripts expressed in the mouse.
Over a period of nearly four years, a method was developed to construct an equalized cDNA library, where cDNA species were nearly equally represented [16]. Nearly 400 cDNA clones were manually sequenced to demonstrate that the normalization technique reduces the presence of abundant cDNA species, resulting in one of the first publications of a large number of cDNA clones analyzed by single-pass sequencing. The data were presented as a table, but the individual sequence was not deposited to the public database, because at that time only high-quality multi-pass sequences were accepted in Genbank. (This rule was changed when Craig Venter's group published their first EST paper, reporting 356 single-pass cDNA sequences as ESTs, as a demonstration of the method's applicability to the genome project [5].) At the same time, we devised a method for high-throughput mapping of cDNAs to the mouse genetic map by PCR, which was presented at the Cold Spring Harbor Laboratory's Genome Mapping and Sequencing Meeting in 1992 and published in 1993 [17,18]. Subsequently, we made a more comprehensive equalized cDNA library (‘whole mouse cDNA catalog’) starting from embryos of all developmental stages [19].
We then made a conventional cDNA library from the micro-dissected extra-embryonic tissues of E7.5 mouse embryos, obtained 3186 ESTs, mapped 155 ESTs on the mouse genome, and reported the clustering of co-expressed genes in the mouse genome, particularly in the t-complex [13]. Using a PCR-based cDNA library construction method [16], we then generated stage-specific cDNA libraries from pre-implantation embryos, at various stages from unfertilized eggs to blastocysts, and obtained around 3000 ESTs from each library [20]. Based on EST frequency analysis, we identified genes that show spike-like transient expression at a specific stage of pre-implantation development. Based on this new finding, we speculated that these stage-specific genes could be key developmental genes and may indeed drive pre-implantation development. We have localized 798 genes from this collection on the mouse genetic map and observed similar trends in the map location of genes, i.e., the clustering of co-expressed genes [20]. In addition, a large number of genes from these collections have been mapped to the Radiation Hybrid map of the mouse genome by the group of Paul Denny and Steve Brown at the MRC UK Mouse Genome Center [21].
3 Current status of the NIA mouse cDNA project
To date, the NIA cDNA project has generated 224 511 high-quality, trimmed ESTs, from 50 individual libraries. One major difficulty in constructing a cDNA library from early embryonic materials is the scarcity of the starting materials. We have recently developed a novel linker-primer design that allows one to differentially amplify long tracts (average 3.0 kb with size ranges of 1–7 kb) or short DNAs (average 1.5 kb with size ranges of 0.5–3 kb) from a complex mixture [22]. The method allows for the generation of cDNA libraries enriched for long transcripts without size selection of insert cDNA. All of our recent cDNA libraries have been made by this new method, and thus, a significant fraction of these cDNA clones contain complete open reading frames, and can be considered near ‘full-length’ clones. Over the past three years, long-insert library construction has produced approximately 140 000 ESTs from early mouse embryos and mouse stem cells (http://lgsun.grc.nia.nih.gov/cDNA/cDNA.html).
Although our EST collection efforts have been focused on early development and stem cells, our cDNA clone sets contain a wide variety of genes that play roles in all types of cellular functions, structures, and biochemical pathways. When the genes represented in our clone sets are categorized by GO annotation [23] (Table 1), they appear to contain a broad cross-section of biological processes, from development to behavior. The proteins encoded by these genes are distributed throughout the cell, membrane, and extracellular space, and they catalyze and/or regulate a wide variety of biochemical functions, from transcriptional regulation to signal transduction. The GO annotation shown in Table 1 is very general, considering only the top two levels of each GO ontology, and only those categories containing a significant proportion of the annotated genes, but it makes the point that in making libraries from a focused set of related tissues, we have not excluded any major types of genes. Considering that over 70% of genes were not classified in each ontology, and the fact that many of the clones were not assigned a UniGene ID [10], it is likely that NIA cDNA clones cover more GO categories than those identified in this analysis. More detailed GO annotation is available at (http://lgsun.grc.nia.nih.gov/cDNA/cDNA.html).
Distribution of NIA cDNA clones in GO categories. UniGene IDs were assigned to the NIA 15K and NIA 7.4K cDNA clone sets, based on clone membership in UniGene clusters, and GO category counts were generated using NIAID's DAVID database [34]. Biological process and cellular component categories from the two upper levels of these ontologies containing at least one percent of the annotated genes were included in the table, while categories included from the more numerous and populated molecular function ontology were limited to those containing at least five percent. Percentages of annotated genes are shown for the NIA 15K and NIA 7.4K cDNA clone sets combined, as well as the number of unique genes, both classified and unclassified in each GO ontology
| Biological process ontology | 15K | 7.4K | combined | ||
| behavior | 16 | 8 | 22 | 0.5% | |
| biological_process unknown | 273 | 134 | 372 | 8.8% | |
| cellular process | 1439 | 783 | 2047 | 48.5% | |
| cell communication | 495 | 342 | 769 | 19.6% | |
| cell death | 90 | 42 | 124 | 3.2% | |
| cell differentiation | 34 | 17 | 47 | 1.2% | |
| cell growth and/or maintenance | 977 | 466 | 1331 | 33.9% | |
| cell motility | 37 | 38 | 65 | 1.7% | |
| development | 293 | 180 | 433 | 10.3% | |
| embryonic development | 23 | 13 | 30 | 0.8% | |
| morphogenesis | 164 | 118 | 259 | 6.6% | |
| pattern specification | 19 | 8 | 26 | 0.7% | |
| regulation of gene expression, epigenetic | 13 | 7 | 20 | 0.5% | |
| reproduction | 34 | 16 | 46 | 1.2% | |
| physiological processes | 2574 | 1233 | 3500 | 83.0% | |
| death | 91 | 42 | 125 | 3.2% | |
| homeostasis | 23 | 10 | 29 | 0.7% | |
| metabolism | 1840 | 867 | 2485 | 63.2% | |
| response to endogenous stimulus | 64 | 37 | 88 | 2.2% | |
| response to external stimulus | 128 | 81 | 196 | 5.0% | |
| response to stress | 131 | 66 | 177 | 4.5% | |
| unique ids | 10 693 | 6468 | 16 302 | ||
| level 1 | total classified | 3062 | 1531 | 4217 | 25.9% |
| total unclassified | 7631 | 4937 | 12 085 | 74.1% | |
| level 2 | total classified | 2852 | 1426 | 3930 | 24.1% |
| total unclassified | 7841 | 5042 | 12 372 | 75.9% | |
| Cellular component ontology | 15K | 7.4K | combined | ||
| cell | 2690 | 1316 | 3688 | 85.1% | |
| cell fraction | 64 | 38 | 98 | 2.4% | |
| intracellular | 2015 | 863 | 2624 | 64.5% | |
| membrane | 1064 | 635 | 1581 | 38.9% | |
| cellular_component unknown | 270 | 130 | 366 | 8.4% | |
| extracellular | 615 | 397 | 947 | 21.9% | |
| extracellular matrix | 72 | 49 | 104 | 2.6% | |
| extracellular space | 590 | 371 | 902 | 22.2% | |
| unlocalized | 30 | 15 | 41 | 0.9% | |
| unique ids | 10 693 | 6468 | 16 302 | ||
| level 1 | total classified | 3134 | 1564 | 4332 | 26.6% |
| total unclassified | 7559 | 4904 | 11 970 | 73.4% | |
| level 2 | total classified | 2943 | 1467 | 4069 | 25.0% |
| total unclassified | 7750 | 5001 | 12 233 | 75.0% | |
| Molecular function ontology | 15K | 7.4K | combined | ||
| binding activity | 2063 | 1045 | 2822 | 59.0% | |
| metal ion binding activity | 274 | 173 | 406 | 9.3% | |
| nucleic acid binding activity | 850 | 360 | 1093 | 24.9% | |
| nucleotide binding activity | 666 | 360 | 935 | 21.3% | |
| protein binding activity | 473 | 254 | 654 | 14.9% | |
| enzyme activity | 1454 | 722 | 1992 | 41.6% | |
| hydrolase activity | 580 | 291 | 793 | 18.1% | |
| kinase activity | 274 | 163 | 392 | 8.9% | |
| oxidoreductase activity | 188 | 85 | 259 | 5.9% | |
| transferase activity | 477 | 263 | 670 | 15.3% | |
| molecular_function unknown | 265 | 133 | 360 | 7.5% | |
| signal transducer activity | 331 | 236 | 532 | 11.1% | |
| receptor activity | 215 | 160 | 348 | 7.9% | |
| structural molecule activity | 219 | 83 | 274 | 5.7% | |
| transcription regulator activity | 257 | 155 | 379 | 7.9% | |
| transcription factor activity | 204 | 122 | 301 | 6.9% | |
| transporter activity | 462 | 209 | 627 | 13.1% | |
| unique ids | 10 693 | 6468 | 16 302 | ||
| level 1 | total classified | 3498 | 1726 | 4787 | 29.4% |
| total unclassified | 7195 | 4742 | 11 515 | 70.6% | |
| level 2 | total classified | 3204 | 1581 | 4388 | 26.9% |
| total unclassified | 7489 | 4887 | 11 914 | 73.1% |
Due to the care taken in preparing these libraries and their resulting high quality, which has been confirmed through sequence verification and sequence analysis, combined with the large amount of clone information publicly available on the NIA Mouse cDNA Project web site, the clones are commonly incorporated into microarrays at many facilities worldwide. Individual NIA cDNA clones are currently available from ATCC.
4 NIA mouse cDNA clone set resources
4.1 NIA 15K Mouse cDNA clone set
The first condensed, non-redundant clone set assembled from our collections was the ‘NIA 15K Mouse cDNA Clone Set’, derived from approximately 53 000 3′-ESTs based on an all-against-all BLAST search [24]. The clone set contains 15 247 cDNA clones representing approximately 12 000 unique transcripts. cDNA libraries used in the assembly of the 15K set include pre-implantation stages (unfertilized eggs, 1-cell, 2-cell, 4-cell and 8-cell embryos, morula and blastocyst [20], micro-dissected tissues of embryonic and extra-embryonic parts of E7.5 embryos [13], female gonad/mesonephros from E12.5 embryos, and ovary from newborn fetus. Approximately 50% of the clones were selected from pre-implantation stage cDNA libraries. Once the condensed clone set was assembled, clone identities were verified by sequencing from both 5′ and 3′ ends [25]. About half of the clones represent transcripts with unknown functions. Approximately 4500 clones with known function have been manually annotated and classified into nine different categories based on their functions as reported in the literature. Information for each cDNA clone in the 15K set is available at the NIA Mouse cDNA Project web site (http://lgsun.grc.nia.nih.gov/). The 15K clone set is available without restriction, and has been distributed to 10 academic centers for further distribution to over 200 research centers world-wide.
4.2 NIA 15K mouse cDNA microarray
Ideally, a cDNA microarray should contain probes representing all of the genes encoded in the genome, but cDNA clones collected by most EST projects are limited to adult tissues so that genes expressed uniquely in early embryos, key genes playing important roles in early embryogenesis, are not included in most available cDNA clone sets and microarrays. Our research group has been working to address this problem by incorporating our specialized gene content into microarray platforms well-suited for the study of early mammalian embryonic development.
The NIA 15K cDNA Microarray is based on the 15K clone set and was first applied to expression profiling of mid-gestation mouse embryo and placenta [24]. This study identifies 720 transcripts as differentially expressed between embryo and placenta, and many of the placenta-specific transcripts identified were related to growth hormone, hormone secretion, and known transcription factors expressed in placenta. We have subsequently applied the cDNA microarray to various expression profiling experiments, such as a comparison of normal and cloned mouse placenta [26], and embryo-derived stem cells such as embryonic stem cells and trophoblast stem cells [27].
4.3 NIA 7.4K mouse cDNA clone set
Recently, we completed assembly of the NIA 7.4K Mouse cDNA Clone Set [28], a non-redundant collection of cDNAs which are not represented in the 15K clone set, as a complementary expansion of the existing gene catalog and microarray. It is comprised of cDNAs collected from embryonic tissues (E0.5 to E12.5), as well as the following stem cells: embryonic stem (ES) cells, trophoblast stem (TS) cells, mesenchymal stem (MS) cells, neural stem (NS) cells, hematopoietic stem (HS) cells, and embryonic germ (EG) cells, with an average insert size of 2.5 kilobases. Preliminary evidence suggests that many of these clones contain full-length inserts. These clones were originally condensed from approximately 11 000 parental clones down to the present 7400 by excluding redundancies within the 7.4K as well as those across the existing NIA 15K library. In an effort to ensure purity and to prevent contamination, the entire 11K parental clone set was single-colony isolated into individually labeled, capped tubes. Re-arraying to the 7.4K clone set was conducted within these tube racks by simply rearranging the tubes in their frozen state and copying the racks into 96-well micro titer plates. These plates were then re-sequenced and clones that were unverifiable, redundant within the set, or overlapping with the NIA 15K set were discarded. To date, the NIA 7.4K Mouse cDNA Clone Set has been transferred to 10 distribution centers worldwide, with additional centers to be added soon, and clones will be made available without restriction.
4.4 NIA 22K 60-mer oligonucleotide microarray
The NIA 15K clone set is in use at microarray facilities around the world, and we sought to expand this resource to incorporate the gene content of the NIA 7.4K cDNA clone set. New microarray technologies reduce the amount of time and labor required to produce microarrays and increase design flexibility [29,30], requiring only sequence information as input. We decided to produce the expanded microarray using an ink-jet based process that synthesizes 60-mer oligonucleotide probes in situ [30] to produce the expanded microarray. We were able to incorporate probes for almost 22 000 transcripts from the NIA 15K and NIA 7.4K cDNA clone sets [31], and begin using the expanded microarray over six months before the expanded clone set was re-arrayed.
Collections of cDNA clones corresponding to microarray features are essential for techniques used to validate and expand on the results of microarray studies, such as northern blotting, in situ hybridization, over-expression, and small interfering RNA (SiRNA) studies. The NIA 15K and NIA 7.4K condensed cDNA clone sets contain clones corresponding to over 98% of features on the NIA 22K 60-mer oligonucleotide microarray, with public access to associated bioinformatic data.
While the gene content of the NIA 22K 60-mer oligonucleotide microarray is broad enough for general expression profiling needs (Table 1), we compared it with that of the Affymetrix MG-U74v2 mouse genome microarray to illustrate the benefits of its specialized gene content, using publicly available UniGene annotation information [32–34] (Table 2). While the Affymetrix platform contains many more total features (36 767 vs. 21 939), it contains fewer unique genes with UniGene identifiers (13 489 vs. 16 600). Furthermore, the Affymetrix platform appears to contain more redundancy, with 23 977 UniGene-identified probes representing only 13 489 unique genes, compared to the NIA 22K platform's 19 195 probes for 16 600 unique UniGene-identified genes. Over 50% of the UniGene-identified genes on the NIA 22K microarray are not found on the Affymetrix mouse genome platform – this group is likely to contain genes which are specific to the early developmental tissue and stem cell libraries used to construct the microarray.
Content comparison of the NIA 22K 60-mer oligonucleotide and Affymetrix MG-U74v2 mouse genome microarrays. Probes from both microarray platforms were assigned UniGene IDs, taken first from publicly-available annotation files [32,33], and second from the NIAID DAVID database [34]. Annotation data was cross-referenced in Microsoft Excel to determine how many probes and unique genes from each microarray platform were also found on the other. Over 9400 unique genes were found only on the NIA 22K 60-mer oligo array, suggesting that the specialized libraries used to build its content have resulted in a large proportion of unique, specialized gene content on this microarray
| UniGene ID | matched | ||||
| NIA 22K | total | + | − | + | − |
| probes | 21 939 | 2744 | 19 195 | 9061 | 10 134 |
| unique genes | – | – | 16 600 | 7113 | 9487 |
| UniGene ID | matched | ||||
| Affy MG-U74v2 | total | + | − | + | − |
| probes | 36 767 | 12 790 | 23 977 | 13 858 | 10 119 |
| unique genes | – | – | 13 489 | 7113 | 6376 |
We have optimized and validated labeling and hybridization protocols for the 60-mer oligonucleotide system for total RNA samples as small as 2 ng [31], to enable microarray studies of early embryos and laser capture microscopy samples. In practice, we have successfully used RNA equivalent to 18 unfertilized eggs in expression profile comparisons (data not shown).
The NIA 22K 60-mer oligonucleotide microarray combines the following unique features and technical advantages to form a powerful system for genome-scale gene expression studies of mouse development: (i) gene content enriched for genes relevant to studies of mouse embryogenomics [4], derived primarily from stem cells and early embryos; (ii) public availability of cDNA clones corresponding to over 98% of features on the microarray; (iii) the ease-of-use and flexibility of in-situ oligo synthesis technology allowing customization and rapid transfer of the platform to other laboratories; (iv) 60-mer oligonucelotide probes which confer greater sensitivity than 25-mers [30], with greater specificity than cDNA probes, resulting in higher differential expression detection rates; and (v) compatibility with reduced amounts of input RNA, allowing its application to early embryos, FACS-purified cells, and microdissected tissues.
5 NIA mouse gene index
Non-redundant clone sets can provide a catalog of transcripts expressed in the tissues which they were collected from, but even when they are as well-characterized as the NIA 15K and 7.4K clone sets, sequence and similarity search information can only partially describe the genes from which those transcripts are derived, particularly in the case of novel or uncharacterized genes. To describe the organization of transcripts within each gene, gene structure, and location within the genome, linkage to higher-level data is required. In an effort to integrate our existing data on clone sequence, similarity searches, and links to other databases with information from the mouse genome assembly (Ensembl), a large set of fully-sequenced cDNA clones [35], and curated gene models (RefSeq), we have created the NIA Mouse Gene Index (Sharov et al., in preparation).
To describe the process briefly, all EST sequences in our collection were re-trimmed using very stringent criteria [36] and filtered to remove undesirable sequence. This pool of ESTs was then clustered using multiple algorithms [37], and the resulting clusters were assembled with RefSeq records, providing very reliable gene identification for over 11 000 assemblies. Furthermore, EST clusters and non-clustering ‘singleton’ ESTs were aligned with Ensembl transcripts, Riken clones, or genomic contigs, and many unidentified EST transcripts were divided into exons on the genomic sequence, suggesting that they are in fact transcripts expressed from those locations in the mouse genome (Sharov et al., in preparation).
Information from numerous other analyses was combined to describe the clone set and the individual transcripts it represents in more detail than ever before. Overall, we estimate that our clone collections represent at least 15 000 unique genes. They contain many clones that extend existing gene/transcript models, and many clones that may represent transcripts specifically expressed in early embryos and stem cells. The information from these analyses will be available at the NIA Mouse cDNA Project web site (http://lgsun.grc.nia.nih.gov).
6 Future directions
Current efforts in the NIA cDNA project are focused upon distributing the 7.4K clone set as well as providing long-insert clones to the Mammalian Gene Collection (MGC) project [38] and American Type Culture Collection (ATCC), as well as to other world-wide distribution centers. In addition, we are currently focused on generating new long-insert cDNA libraries from later stages of mouse embryo development.
Acknowledgements
The authors would like to thank the many collaborators who have contributed tissues and RNA samples to the NIA Mouse cDNA Project, which are too numerous to list here. We would also like to stress that this manuscript is not intended to be a comprehensive and thorough review of mouse genomics literature, but rather an account of our research group's work in the field. There are numerous works not cited here, due to space limitations and the focus of this manuscript, and we offer our apologies for any such omissions.