

1 Introduction
At the beginning of the new millennium, it might be difficult to realize that, in the early 1950s, the nucleic acid metabolism held very peripheral interest, because so little was known about it. The pathways of sugar metabolism, the Krebs cycle, the hydrogen transport systems, enzyme kinetics, etc. were much more on focus and benefited from a much greater emphasis.
Major breakthroughs occurred in the middle of this background. They came from the 1953 discovery of the DNA double helix by J.D. Watson, F. Crick, M. Wilkins and R. Franklin, but also from the demonstration that nucleic acid-like polymers could be synthesized in vitro, following the characterization of the enzyme, PNPase, by M. Grunberg-Manago and S. Ochoa. Similarly, the possibility to replicate DNA in vitro, as discovered by A. Kornberg, constituted an important milestone of molecular biology.
It was only some years later that people realized that PNpase does in fact play a degradative rather than biosynthetic role in vivo, and that DNA polymerase I is involved in repair mechanisms rather than replication properly said, a biological process that mobilizes a large number of proteins. But the mere fact that nucleic acids, or nucleic acid-like macromolecules, could be, either synthesized, or copied from pre-existing templates, using a cell-free system, gave a real impetus to molecular biology, and to the elucidation of the genetic code.
These early studies were contemporary to the notion that DNA is, in the cell, the template for RNA synthesis, and it was much to the merit of people like S. Weiss, J. Hurwitz, and L.C. Stevens, to have shown, for the first time, the possibility to synthesize RNA in a DNA-dependent manner, using a crude cell-free system. Soon afterwards, they succeeded in purifying the enzymatic entity responsible for this reaction and gave it the name of (DNA-dependent) RNA polymerase (later called ‘transcriptase’). Subsequently, P. Chambon and co-workers made major contributions in this field by establishing the existence of distinct RNA polymerases endowed with different specificities regarding the types of RNAs to be transcribed.
2 The messenger RNA ‘saga’
The year 1961 marked the beginning of what I am designating here as the ‘messenger saga’, namely the messenger hypothesis, derived mainly from J. Monod and F. Jacob's studies on the kinetics of enzyme induction, and the direct identification of messenger RNA in phages and bacteria. This had been preceded by some remarkable, albeit intriguing observation due to A. Volkin and L. Astrachan, who had characterized, in 1958, a peculiar RNA fraction that was synthesized during T2 phage development, the base composition of which was quite similar to that of T2 phage DNA.
The identification of messenger RNA (S. Brenner, F. Jacob, and M. Meselson, F. Gros et al.) as well as the cardinal observation by B. Hall and S. Spiegelman that RNA from phage-infected E. coli cells can form an heteroduplex with the DNA of that phage lent support to the ‘central dogma hypothesis’ (F. Crick) according to which the genetic information is unilaterally and irreversibly channelled from DNA to RNA and from RNA to proteins. That nature can exhibit some exceptions to this rule resulted, as is well known, from H. Temin and D. Baltimore's discovery (1967) that retroviruses can transcribe RNA into DNA.
Concerning the existence of restriction enzymes (which turned out to become the main technical ‘weapons’ in the field of genetic engineering), I remember an early lecture given by W. Arber, in Paris, at the ‘Club de physiologie cellulaire’ in the presence of J. Monod, F. Jacob and others. Although the data were convincing and the lecture well argumented, everyone regarded the restriction–modification phenomenon as illustrative of a peculiar strategy utilized during the phage–bacterium interaction, while paying little attention to the types of endonucleases involved. People's attitude regarding the importance of these enzymes changed drastically with the fantastic development of the recombinant DNA technology.
2.1 The first cDNA cloning experiments
The first evidence and the first demonstration that cDNA could be cloned in a bacterial plasmid came from work by F. Rougeon, P. Kourilsky and B. Mach in 1975, that was published in Nucleic Acids Research [1]. They showed that double stranded DNA made by reverse-transcribing rabbit globin messenger RNA could be recombined in vitro with a pBR22 plasmid DNA, and the recombinant thus obtained used to transfect E. coli cells, enabling one to further isolate clones of globin cDNAs. This was soon followed by similar work by the groups of Rabbitts [2] and Maniatis [3].
The recombinant DNA technology later resulted in a wealth of experiments giving rise to the cloning of many eukaryotic genes including some early examples obtained at the Pasteur Institute on chicken ovalbumin [4], mouse immunoglobulin chains [5–8], human growth hormone [9], mouse histocompatibility antigens [10], mouse rennin [11] and the acetylcholine receptor alpha subunit of Torpedo marmorata [12].
2.2 Capping, polyadenylation, splicing and transcriptional control
Major observations were made between 1976 and 1978 about the physiological modifications of eukaryotic RNA transcripts. For example, it was shown by G. Brawerman and U. Littauer that, in most cases, eukaryotic messengers are polyadenylated at their 3′ ends. Although the role of this poly-A tail is not fully understood, specific poly-A binding proteins have been described and it has been postulated that they could be responsible for the extra nuclear transport of primary transcripts and for controlling their metabolic stability. At the 5′ end, another type of modification taking place is the phenomenon of ‘capping’ consisting upon a terminal guanylation by a guanylyl-transferase, plus a methylation of the guanine residue by a 7-methyl transferase, as shown initially independently by Lewin and Furuichi [13]. This guanine-rich cap is believed to exert some transient protection of RNA transcripts and to prevent them to adopt a spurious secondary structure.
But what came out as the greatest surprise – particularly to molecular biologists trained in microbial and phage genetics – was the discovery of ‘split genes’ by P. Sharp and the observation by many others, including P. Chambon, that the product of these genes are long transcripts that have to be processed (spliced), according to a mechanism of exon shuffling, in order to give rise to mature messenger RNAs. The existence of self-splicing mechanisms operating in the excision of particular introns from Tetrahymena ribosomal RNA (T. Cech), or from plant viral RNAs, further led to the exciting concept of catalytic RNAs, so called ‘ribozymes’, a concept which has had considerable implications in the field of molecular evolution, and which is finding recent support in the manifestations of the so called ‘RNA world’ (interfering RNA, riboswitches, non sense-mediated mRNA decay, etc., see for example [14–18]).
But, between 1980 and 1985, molecular and developmental biologists turned most of their attention to the control of eukaryotic transcription itself. Among the names that have best illustrated this field in its early phase one should mention people like R.G. Roeder, P. Chambon, M. Yaniv, A. Klug, etc. The main message that came out from their work was that, contrary to the situation observed with prokaryotes, gene regulation in eukaryotes obeys, in most cases, a positive rather than negative control, which is achieved by the attachment of transactivating factors to cis-acting regulatory sequences lying upstream of the transcription start-site. As illustrated in Fig. 1, showing as an example the desmin gene control region, a three dimensional arrangement of these different factors has to be taken into consideration to understand how they operate to stimulate the activity of the RNA polymerase. The role of ‘enhancers’ presumably involved in this type of interaction at the chromatin level was shown to be a very crucial one.
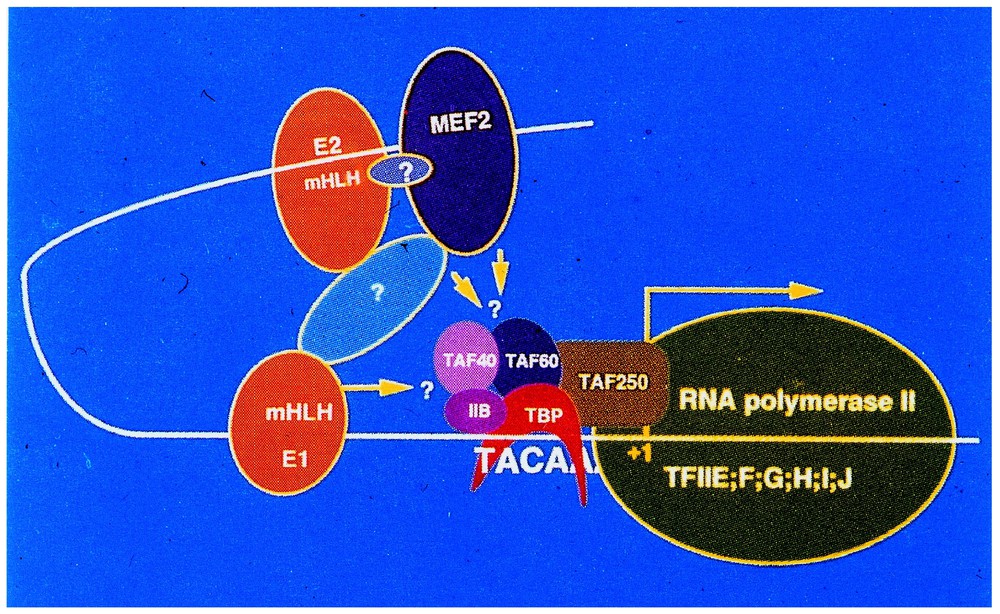
Schematic representation of the Desmin-gene control region (courtesy of Dr. D. Paulin, University Paris-7). mHLH E1 and E2: HLH myogenic regulatory factors with specific affinity to the E-box; MEF-2: myogenic trans-activating factor; TAF40, 60, 250 TFIIB, TBP, ubiquitary transactivating factors; TFIIE, F, G, H, I, J: TATA-box binding proteins.
3 The transcriptome era
3.1 The transcriptome and the utilization of microarrays
It is clear that the technical analysis of cellular transcripts went through different phases some of which consisting of crude approaches to global cell sub-fractions, others being more sophisticated, including fractionation of mRNA by sucrose gradient ultracentrifugation or preparative electrophoresis [19]. For example, and as already mentioned, bacterial messenger RNA was initially defined as a rapidly labelled, heterogeneous, and metabolically unstable RNA fraction capable of reversible attachment to ribosomes. In other instances, its existence was also inferred from its capacity to stimulate protein synthesis in a reconstituted cell-free system. Later on, following work on RNA polyadenylation, the messenger subfraction from eukaryotic cells could be purified by chromatography on oligo-dT-cellulose columns. Then radioactive cDNA became available, the PCR amplification technique came to age, and more sophisticated methods could then be used. Instead of addressing messenger RNA as a global component of the cell, it then became possible to deal with gene-specific transcripts. Combining detection with labelled cDNA probes and electrophoretic separation, the ‘Northern blot’ analysis greatly facilitated the identification of specific gene transcripts in cell-free extracts.
More recently, with the onset of genomics, new technologies have been developed that give the possibility to monitor the expression of hundreds or thousands of genes at a time. These technologies are essentially based on the utilization of high-density microarrays (DNA chips). They usually involve hybridization of total cDNA samples derived from the whole collection of RNA transcripts present in a cell extract, to genes or to oligo-chips immobilized on solid supports such as nylon membranes or glass slides.
Today, using oligonucleotides, cDNAs or gene fragments present on these supports, it is possible to have access to the whole set of messenger RNAs that are expressed by the cell or the tissue under precise physiological or developmental conditions. This complex collection of transcripts is designated under the name of ‘transcriptome’. It is the ‘signature’, so to speak, of the total gene expression pattern of the cell at any given moment of its normal or pathological fate.
Recent advances in different technical fields such as material engineering, optics, electronics, robotics, chemistry, genetic engineering and informatics have permitted the development of these miniaturized platforms, the utilization of which is extremely versatile. They can be used to help biologists to scrutinize the cell physiology in general. For example, transcriptional regulatory circuits, cell division patterns, apoptosis, communication and signal transduction networks, etc., all of these parameters can influence gene expression in such a way as to affect the transcriptome.
The microarray techniques can also afford new tools for diagnosing diseases, for establishing cancer prognosis, for assisting the field of drug development (pharmacogenomics), or else to tailor therapeutics to specific pathologies, and, of course, to generate databases.
There exist several types of microarrays [20] (Fig. 2). Sometimes the oligonucleotides serving as probes for detecting cell transcripts or their cDNAs can be synthesized directly in situ, by photolithographic techniques (Affymetrix); the synthesis takes place on glass support and up to half a million probes can actually be synthesized on a very small surface (1.3 cm2). The same can be realized by an ink-jet system. On the other hand, pre-fabricated oligos or cDNAs can be printed on glass or Nylon supports by robots; this procedure was utilized to analyse gene expression in yeast, since close to 5–6000 genes from this organism could be deposited on a surface not exceeding 2 cm2.

Different types of micro-arrays. (a) Oligonucleotide array synthesized in situ with photochemical technology by Affymetrix; (b) oligonucleotide array synthesized in situ with inkjet technology (image courtesy of Rosetta Inpharmatics); (c) DNA micro-array printed on a glass slide (image courtesy of Corning, Inc.). (Reproduced from [20].)
3.2 Gene-expression profiling and cancer
DNA arrays are also quite utilized by physicians or research scientists confronted with the nosology (categorization) and prognosis of various types of cancers.
For example, Lander and Golub [21] succeeded in establishing interesting comparisons at the transcriptome level between acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL), two types of blood diseases that are not always easy to distinguish in their early stages using classical approaches. Gene profiling study of the RNA transcripts from 40 leukaemic patients (with micro-arrays containing 7000 human gene probes) could show that up to 50 genes were differentially expressed in AML and ALL.
The same type of approach was applied at Stanford by Brown and Botstein [22] in analysing the development of diffuse non-Hodgkin lymphoma. Using specific arrays containing up to 18 000 genes, they were able to observe distinctive traits between low-risk and relatively high-risk lymphomas. But blood cancers are not the only type of cancers that are amenable to comparative transcriptome analyses. Work done on melanomas [23–25] pointed out to the major role played by the Rho-C, Thymosin β-4 and fibronectin genes. High-level expression of these genes was found to be correlated with bad prognosis, namely conversion of local subcutaneous tumor cells into metastatic ones. Rho-C is a small GTPase, which functions as a mitogenic factor and is probably indirectly involved in cell adhesiveness. Thymosin β-4 exerts some direct effects on the acto-myosin containing filaments, making the active monomers more available to rapid polymerisation into lamellipodia. Fibronectin is a component of the extracellular matrix, the deposition of which favours the movement of the cell on a surface. Microarray studies have permitted to include these three components in the acquisition of the metastatic state by melanomas.
Much work has also been devoted, using similar approaches to the prognosis of cancers affecting breast (Fig. 3) [26], ovaries, lung and prostate with interesting results [27–29].
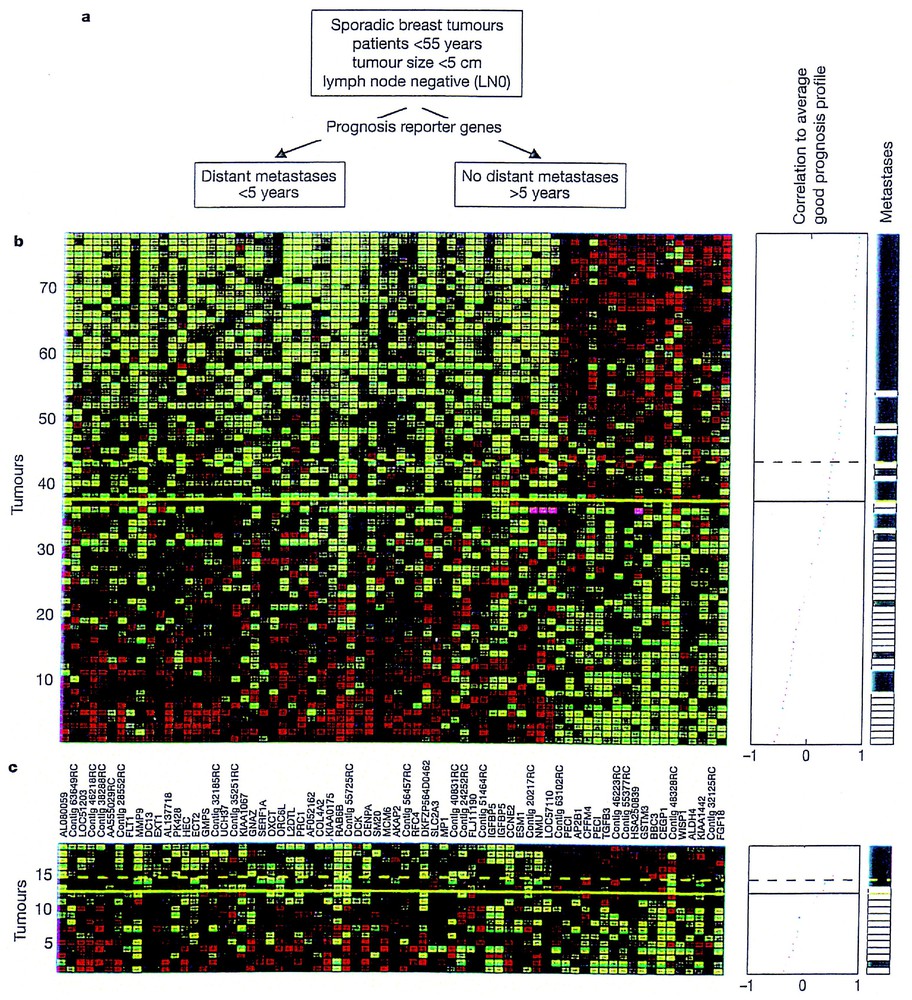
Gene expression profile and breast cancer prognosis. (a) Use of prognostic reporter genes to identify two types of diseases outcome from eight sporadic breast tumours into a poor prognosis (distant metastasis in less than five years) and good prognosis group (no distant metastasis) observed after five years. (b) Expression data matrix (the authors are using 70 prognostic marker genes – each column represents a gene – from tumours derived from 78 breast cancer patients – each row represents a tumour). Genes (whose names are indicated between (b) and (c)) are ordered according to their correlation coefficient with the two prognostic groups. Tumours are ordered by the correlation to the average profile of the good prognosis group (middle line). Middle panel: above the dashed line patients have a good prognosis signature, below the line, the prognosis is poor. Right panel: indicates the metastasis status of the patients; white indicates who have developed distant metastasis within five years after early diagnosis; black corresponds to patients who continued to be disease free for at least five years. (c) Same as for (b), but corresponds to the expression data matrix for tumours of 19 additional breast cancer patients (reproduced from [27]).
4 Concluding remarks
Over the last 40 years, considerable progress has been made in our way to identify and characterize the gene transcription products of the cell under various physiological conditions. The study of the cell transcription pattern has begun with the early characterization of global messenger RNA fraction, using tedious sucrose gradient or oligo-dT cellulose separation techniques.
The second phase coincided with the availability of cloned cDNAs. It made possible to test particular hypotheses in relation to the activity and control of specific genes. With the development of genomics, new technologies have emerged that permit to observe and analyse global tissue-specific expression profiles in a single step. Interestingly, the microarray approach is opening a new era for it does not involve any a priori hypothesis in scrutinizing the transcriptional activity of a cell. Rather, the biologist is in the position to examine which genes are overexpressed or repressed, sometime without even knowing their function. This can orient the experimentalist towards particular ‘clusters’ of genes that evolve in a coordinate fashion, with the view to further explore whether their expression results or not from an integrative regulatory circuit.
It seems nonetheless to me that, with the study of transcriptomes, we are in about the same situation as were the Egyptologists from the 19th century. We find ourselves at the very beginning in our ability to decipher the various signatures of the cell! But there is little doubt that, in the years to come, and with the help of bioinformatics, the analysis of transcriptomes (as well as that of the proteomes) will revolutionize our understanding of cell physiology, while becoming central to the development of functional genomics, and to some important medical approaches.