

1 Introduction
Because of immovability, the plants are inevitably affected by various environmental factors in the process of evolution [1–18]. Plants are remarkably sensitive to environmental conditions and have evolved the ability to undergo adaptive developmental and physiological changes in response to environmental stimuli [19–26].
The sound wave, as a form of alternative mechanic stress, has obvious effects on the growth and development of plants [1,2]. It was reported that some sound wave could accelerate the growth of plants and change the transition temperature of cell walls that were tightly related to the division of plant cells [3]. In the study of the relation of sound wave and deformability of plasmalemma [4], it was found that the deformability was close to the frequency of the sound, and with the increase of frequency, the deformability decreased. Furthermore, the study of the effects of sound stress on nucleic acid and soluble proteins shows that the content of DNA did not change evidently, that of RNA and soluble proteins increasing with varying degrees. The genes in response to sound are started up under sound stimulation [5]. However, the started-up genes sensitive to sound stimulation are not clear [1,22–26].
In the present work, we used DDRT-PCR to isolate the differentially expressed gene fragments responding to sound-wave stress on Chrysanthemum. The relation of plant growth and development and sound-wave stress was also discussed, which would provide a solid basis for investigating molecular machinery of flower plant adaptation to environmental stresses [20–25].
2 Materials and methods
2.1 Chrysanthemum callus culture
Stems from Chrysanthemum seedlings were inoculated in conical flasks with 20 ml of MS solid medium each and cultured in an illumination incubator at 26 °C [6].
2.2 Sound-stress stimulation and plant material
The sound generator in our laboratory achieved alternating stress. Inoculated stems were stimulated by sound wave with certain intensity (100 dB) and frequency (1000 Hz) for nine days, respectively, and each day for 60 min. The control group was placed in the same environment as stressed groups. The strong sound field apparatus refers to [6]. Fresh leaves of stressed groups and control groups were frozen in liquid nitrogen before the extraction of total RNA, respectively.
2.3 Total RNA extraction
Total RNA was extracted from leaves, as described by Burgermeister et al. [1]. 0.5 g of leaves from several plant materials were pooled for one isolation after purification by phenol:chloroform:isoamyl alcohol (50:49:1, v/v) and ethanol precipitation. RNA was resuspended in a total volume of 100 ul of double-distilled water. Agarose gel electrophoresis was used to test the integrity and a UV spectrophotometer was used to detect the purity of the RNA extracted.
2.4 mRNA differential display
mRNA differential display was carried out as described in the handbook of GenHunter Corporation (1994). Anchored primers (HT11M: 5′-AAGCTTTTTTTTTTTA/G/C-3′) were synthesized by Sangon. In a 0.5-ml microcentrifugation tube were added 2 ug of total RNA, 4.96 ul of 20 uM anchored oligo-dt primer; the mixture was brought to 11 ul with DEPC-treated water. The tube was heated in a water bath at 70 °C for 5 min, and then cooled on ice. 4 ul of 5 M MuLV RT buffer, 2 ul of 10 mM 4dNTP mix, 0.5 ul of 20 U RNase inhibitor, and 1.5 ul of DEPC-treated water were added. The mixture was incubated at 37 °C for 5 min. 1 μl MMLV reverse transcriptase (200 U/μl) was added for 60 min at 42 °C. The reaction was heated in a water bath at 70 °C for 10 min, and then cooled on ice. The products of the reverse transcriptions were stored at 4 °C for immediate use or at −20 °C for later use.
PCR amplification of each reverse transcription products was carried out in combination with one of eight arbitrary primers H-AP (HAP1, AGCTTGATTGCC; HAP2, AAGCTTCGACTGT; HAP3, AAGCTTTGGTCGA; HAP4, AAGCTTCTCAACG; HAP5, AAGCTTAGTAGGC; HAP6, AAGCTTCGACCAT; HAP7, AAGCTTAACGAGG; HAP8, AAGCTTTTACCGC) synthesized by Sangon. PCR was carried out as followed: 94 °C for 30 s; 40 °C for 2 min; 72 °C for 30 s for 40 cycles, and finally 72 °C for 5 min.
The amplified cDNA subpopulations of 30-terminals of mRNAs as defined by this pair of primers were size-fractionated by denaturing in polyacrylamide electrophoresis using Bio-Rad Mini-PROTEAN Electrophoresis Cell. 3.5 ul of each PCR sample were incubated with 2 ul of loading dye (95% formamide, 10 mM EDTA, pH 8.0, 0.01% bromophenol blue) at 80 °C for 2 min immediately before loading.
2.5 Silver staining
Silver staining of the polyacrylamide gel was performed as described by the Bio-Rad Silver Stain Handbook, with the modification that the gel was fixed using 7.5% acetic acid (v/v) immediately after electrophoresis; the developmental reaction was stored using 7.5% acetic acid (v/v).
2.6 Re-amplification and northern blot hybridization
The gel band of differentially expressed cDNA fragments was cut down with a knife and added to an Enppendorf tube in which it was boiled for 15 min. The extract served as the template to re-amplify with the same pair of primes as in PCR.
2.0 ug of total RNA from the treated and the control samples were denatured at 70 °C for 5 min, and spotted onto a nylon membrane (Hybond-N+, Amersham, UK). The RNA on the membrane between two sheets of filter paper was fixed in an oven at 80 °C for 2 h. Probe-labelling and hybridization conditions were described in the instruction of the northern and southern direct HRP labelling and detection kit (Pierce, USA). The exposure time was 30 min.
3 Results
We made use of the total RNA extraction method described; 200 μg of total RNA were routinely obtained from 1 g (FW) of the callus of Chrysanthemum (Table 1). Results of RNA isolation from the sound stimulating samples and the control samples were 201.3 μg/g and 198.6 μg/g as the mean of five samples, respectively. The A260/A280 ratio ranged between 1.84 and 2.01, indicating little or no protein contamination. The RNA yield of sound-stimulating samples was more than that of control samples.
After total RNA extraction, 3.5 ul of total RNAs were separated on 1.2% denaturing agarose gels and photographed under UV light. RNA examined by electrophoresis on 1% agarose/TAE gels showed that the lightness of the 28s rRNA band was double-fold than that of the 18s rRNA band, indicating that little or no RNA degradation occurred during the extraction (Fig. 1). In Fig. 1, the RNA bands of the treated group were coloured brighter than those of the control group. The quality of the total RNA could assure the need for subsequencing tests.

Total RNA on 1.2% agarose gel electrophoresis.
Through separating these PCR products on denaturing polyacrylamide gel electrophoresis, six differentially expressed cDNA fragments were obtained, which were SA3, CA2, SG2–1, SG2–2, SG7–1, SG7–2 (Fig. 2). The fragments' molecular weight was between 200 and 600 bp, respectively. After being re-amplified (Fig. 3), the SG2–2 band had not outcome, others had a similar molecular weight on the agarose gel and on the denaturing polyacrylamide gel. SG2–2 was fake positive and discarded. Northern dot hybridization was performed (Fig. 4).
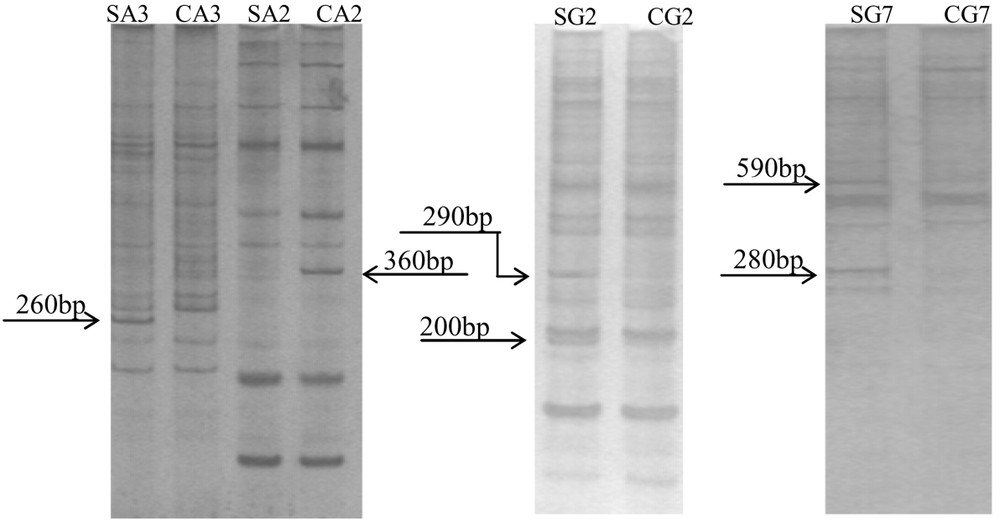
mRNA differential display visualized by silver staining. SA3, SA2, SG2, SG7 are the treated groups; CA3, CA2, CG2, CG7 are the control groups; A3, A2, G2, G7 are respectively the primer pairs of anchored primer and arbitrary primer (HT11A and HAP3, HT11A and HAP2, HT11G and HAP2, HT11G and HAP7).

Agarose gel electrophoresis of re-amplification products. M, molecular size marker.
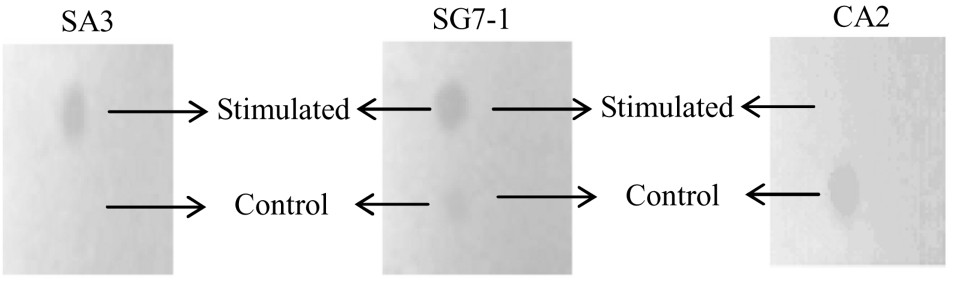
Northern dot hybridization analysis of cDNA fragments.
4 Discussions and conclusions
For the successful isolation of intact RNA from plant tissues, it is important to prevent the contaminating substances from binding to nucleic acids. In our experiments, many difficulties were encountered from the stage of cell lyses to RNA separation in the supernatant and subsequent reactions. The major problem was low yield, degradation, and poor RT-PCR amplification. The success of the RNA isolation procedure may be judged by the quantity, quality and integrity of the RNA recovered [1–7]. The fine quality of RNA isolated as described in this study was confirmed in several ways. First, the A260/A280 absorbance ratio was 1.9–2.0 and the A260/A230 absorbance ratio was 3.1–4.0, indicating that RNA was relatively free of protein and polysaccharide contamination. Second, 1.2% denaturing agarose gel electrophoresis showed clear, discrete ribosomal RNA with no apparent RNA degradation, suggesting that RNA was also relatively free of RNases. Third, the RNA quality was tested by DDRT-PCR. Results showed that, as expected, about 200 and 600 bp DNA differential display fragments were detected in the DDRT-PCR products when RNA was used as a template. RNA was also used for northern analysis.
In Fig. 1, the results demonstrated that the content of RNA increased under sound stimulation. The same result was obtained by Wang [5]. As shown in Fig. 2, a total of six bands were displayed, ranging in size from 200 to 600 bp. Five differential bands were found from stressed group, when control group had one (Fig. 2 indicated the differential bands clearly). To determine true differentially display bands among them, we carried out northern dot hybridization. Three of them were found to be positive fragments, SA3, SG7–1 and CA2, whose molecular weights are 260, 590, and 360 bp, respectively. SA3 was differentially expressed, and SG7–1 was advantageously expressed, while CA2 was restrained by the sound wave. The fact indicated that expression of some genes was turned on; meanwhile, the stress restrained some genes from expression under this mode of sound stress. Wang [7] considered that sound stress have double effects on plant growth, namely, not only can sound stimuli hinder the plant growth, but also sound at a proper frequency and intensity can promote cell division. Our experimental results hold up the view. The three gene fragments acquired will be cloned, and sequenced, and then their functions will be further analyzed in the next experimentation.
In a word, precise elucidation of plant gene regulatory network system under abiotic stresses is of importance to molecularly engineering plant resistance; it is the reason why many excellent scientists worldwide have been engaged in this frontier field, which resulted in a long-step progress [8–26]. One of the greatest challenges is to screen for differentially expressed genes under corresponding environmental stimuli and analyze them, which is the pre-condition for elucidating the molecular mechanism of the phenomenon [9–21]. Performance and change of physiological functions under environmental stresses are phenotypes of potential gene expression underlying this mechanism for flower plants [21–26]. We have established a powerful platform to further solve this hot topic. There are also many issues remaining to be solved and needing to make efforts. The scope of tested plants needs to be extended; comprehensive study on a combination of environmental stress factors in laboratories and in the field should be given much attention; system development viewpoint and computer simulation analysis should also be applied.
Acknowledgements
We thank the National Natural Science Foundation of China (No. 30470431), the Natural Science Foundation of Chongqing (CSTC 2005 BB1094), and the Doctoral Fund of Qingdao University of Science and Technology (0022221).
Vous devez vous connecter pour continuer.
S'authentifier