

1 Introduction
Wheat (Triticum aestivum L.) production is adversely affected by drought in 50% of the area under production in the developing and 70% in the developed countries [1]. As water resources are likely to decline in the coming decades [2], the areas devoted to wheat production will be increasingly threatened by water availability. Hence, improving wheat adaptation to drought will acquire a greater socioeconomic importance across the globe than it currently has.
As shortage of water is more limiting to crop production in arid regions than any other single factor, a better understanding and control of the mechanisms that enable a plant to adapt to low water potentials and maintain the processes involved in growth, development and production, has been an aim of breeding for drought resistance [3]. Therefore, the analysis of the physiological responses of various wheat genotypes to water stress was liable to lead to the development of more efficient selection criteria [4,5].
Whichever adaptation strategy is exerted in the wheat plant as a response to drought, it is paramount to elucidate the central element of control from the atomistic, reductionistic view. The dissection can be conducted at the translational, transcriptional or genetic levels for furthering the manipulation of the components of a genotypic response [6,7]. During the past three decades, 76 genetic and information tools have been developed (and adopted) to reveal basic features in the genetics and expression of different wheat species (Fig. 1). Some are presented as following.
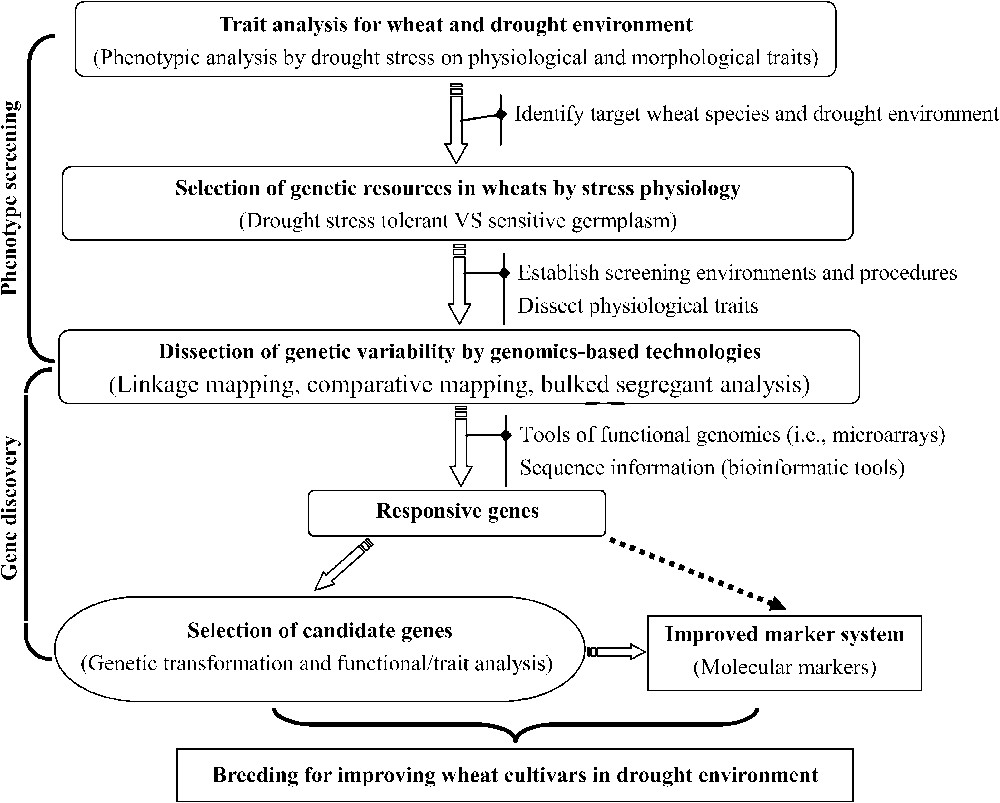
Molecular strategy for increasing drought tolerance uses a top-down approach (phenotype to gene) by beginning with trait analyses in drought environment.
2 Comparative mapping
Comparative maps allow transfer of information about genetic control of traits from species with small diploid genomes, such as rice (Oryza sativa L.), to species with more complex genomic structures (increased repetitive DNA, polyploidy) and less economic support [8,9]. Because of the size and complexity of the genomes, it may not be appropriate to sequence the entire genomes of wheat (Triticum ssp.), rye (Secale cereale L.), oat (Avena sativa L.), or barley (Hordeum vulgare L.) [9,10]. However, alternative strategies involving identification of gene-rich regions of the Triticeae genome and comparison of the genome structure and genetic colinearity with rice, maize (Zea mays L.), sorghum (Sorghum vulgare L.), and other species provide Triticeae researchers with the knowledge and tools necessary for genetic parity with simpler genomes.
Crop species of the Poaceae display a remarkable level of genetic similarity despite their evolutionary divergence 65 million years ago [11]. Molecular markers have been used to develop comparative chromosome maps for several members of the Gramineae and these have been used to study genes of agronomic importance across species [12,13]. Large segments of the genomes of maize, sorghum, rice, wheat, and barley conserve gene content and order [14–17], although the correspondence has been modified by duplications, inversions, and translocations. For the domesticated grasses, the conserved linkage blocks and their relationships with rice linkage groups provides the insight into the basic organization of the ancestral grass genome [18]. This allows the transfer of information from species with small diploid genomes, such as rice, to species with more complex genomic structures, such as that of wheat [16,19].
Despite the progress in comparative mapping, the application of this technology, especially for wheat, rye, oat, and barley will not be realized unless scientifically sound strategies for studying drought tolerance are devised that allow researchers to utilize genetic tools and information developed for model species [20]. This will require more detailed comparative genetic analysis from the DNA sequence of genes all the way to comparative analysis of QTL (quantitative trait locus).
3 Bulked segregant analysis
The usual method to locate and compare loci regulating quantitative traits (QTLs) requires a segregating population of plants with each one genotyped with molecular markers [21]. However, plants from such segregating populations can also be grouped according to phenotypic expression of a trait and tested for differences in allele frequency between the population bulks: bulk segregant analysis (BSA) [21,22]. The same probes used for making a genetic map (e.g., isozyme, RFLP, RAPD, etc.) can be used for BSA [23]. A molecular marker showing polymorphism between the parents of the population and which is closely linked to a major QTL regulating a particular trait will mainly co-segregate with that QTL, i.e. segregate according to the phenotype if the QTL has a large effect [21–24]. Thus, if plants are grouped according to the expression of the trait and extreme groups tested with that polymorphic marker, the frequency of the two marker alleles present within each of the two bulks should deviate significantly from the ratio of 1:1 expected for most populations [23]. As chromosomal locations of many molecular markers have now been determined in many species, the map location of closely-linked QTLs can therefore be deduced without having to genotype every individual in segregating populations [25].
This has been used successfully with composite populations of wheat to locate QTLs associated with yield under severe drought. An inbred line derived from one of the populations selected for higher drought yield has been crossed with a drought-susceptible inbred line to produce a mapping population for QTL analysis of physiological and developmental traits likely to regulate yield under drought [21,26]. Some researchers used bulked segregant analysis (BSA) to identify microsatellite makers associated with water-stress tolerance in wheat, and found that one microsatellite fragment that was present in tolerant parent wheat and the tolerant bulk but absent in the sensitive parent wheat and sensitive bulk [27]. Future work to identify traits having QTLs with flanking markers showing significant allele frequency differences in the BSA studies will indicate that those traits are likely to be important in determining yield under drought.
4 Linkage mapping
The establishment of genetic linkage maps provides the basis for mapping the gene(s) responsible for the expression of traits of interest. In wheat, such maps have also corroborated cytological evidence of major chromosome rearrangements [28–30] and have allowed the comparative mapping among related species [14,31,32].
Within the span of the past thirty years, molecular markers have been considered important biotechnology tools for enhancing the magnitude of plant breeding. From the conceptualization and delineation of perspectives in their use in breeding programmes [33] to their usefulness and efficacy proposals [34,35], the methodologies have ample applications: characterization of genetic diversity [36,37], introgression of exogenous genetic material for diversity increment [33], advancement in novel varieties release [38] diagnostics [39] or selection tools [40].
In several cereal species, genetic linkage maps have allowed the identification of regions controlling some traits related to the response to drought. Different segregating populations from maize, rice, sorghum, barley, durum (tetraploid) wheat and sugar cane (amongst others) have been studied for many different criteria or quantitative characters, such as phenology, plant architecture, metabolic pathways, water-use efficiency or carbon isotope discrimination [41–44]. In contrast, developments in molecular genetics in wheat have been relatively slow and exiguous. Explanations are ample: wheat's ploidy level (, AABBDD); genome size (estimated to contain 18.1 picograms of DNA per haploid nucleus [45], equivalent to ca. pairs); and genomic complexity (>75% consists of repeated DNA sequences of varying degrees of reiteration and length, with a lesser proportion (ca. 20%) of low-copy number or unique sequences [46]).
The wheat hexaploid nature and its amenity to cytogenetic manipulation do offer unique tools to geneticists, allowing them to determine evidence of major chromosome rearrangements [28,29] and the comparison of linkage maps among related species [14,47]. The low number of quantitative traits dissected into their QTL is a reflection of the focus given to simply inherited traits [48,49] and the difficulty of building comprehensive genetic linkage maps for wheat [50–52].
5 Gene discovery
Rapid discovery of genes by large-scale partial sequencing of selected cDNA clones or expressed sequence tags (ESTs) is the initial step towards characterization and categorization of genetically complex abiotic stress responses [53–55]. Expressed sequence tag (EST) analysis was proposed for efficient sampling of a genome for information about genes that could be useful in searching at databases [56]. By searching online databases for similar genes with known function, one can determine if a specific gene (or gene motif) has been found in the same or other organisms and if its function has been determined. These ESTs can also be useful for further laboratory work in gene expression, mapping and direct alteration of the organism [57–59]. EST information can be merged with that of a protein database to provide information on patterns of gene expression. For the long term, EST information will be a critical resource for crop improvement and will be used extensively for locating genes, understanding changing patterns of gene expression, and biotechnological modification of traits. However, identifying and mapping all the expressed genes in a species without sequencing the entire genome is a complex task.
Extensive EST collections and databases already exist for Arabidopsis [60] and rice [61,62], while large-scale EST sequencing initiatives for various crop species is under course [63,64], including that of wheat. However, such collections are biased towards high-to-moderate abundance studies that are derived from different tissues, organs or cells, different developmental stages, various external stimuli, and treatments with plant-growth regulators [65]. In contrast, relatively few studies have focused specifically on ESTs from plants that have been exposed to environmental stresses [65,66].
6 Candidate genes
A candidate gene is such that is associated with the variation in a trait, involved with the development or physiology of the trait. Frequently, candidate genes are sequenced genes of known or suspected function and may belong to biochemical or regulatory pathways [67,68]. Identifying the genes involved in complex trait governance derived from QTL analysis can provide different kinds of genetic information, regularly over a broader range of germplasm. Because there may be large numbers of genes located in the region of a QTL, the odds of identifying the gene that actually controls the expression of the trait appear to be quite low; however, a number of factors can increase the odds of success, especially as the number of genes sequenced increases [69]. Translational genomics of these candidate genes using model plants provided encouraging results, but the field testing of transgenic crop plants for better performance and yield is still minimal [70]. Expression microarrays provide new insights into physiological and biochemical pathways of drought tolerance, and thus can lead to identification of novel candidate genes that can rapidly advance breeding for drought tolerance [71,72].
Geneticists are using association genetics to dissect complex adaptive traits and discover the underlying genes. In parallel, they are using resequencing of candidate genes and modern population genetics methods to discover genes under natural selection. This combined approach is identifying the most important genes that determine patterns of complex trait adaptation observed in many crop populations [73]. In addition, researchers are now routinely using candidate gene-based mapping and genome-wide linkage disequilibrium and association analysis in addition to classical QTL mapping to identify markers broadly applicable to breeding programs.
7 Functional genomics
Using DNA chips [74], it may be possible to determine the relative importance (contribution) of each gene to some of the studied physiological traits involved in drought adaptation at different phenological stages under different water regimes [13,20,75,76]. Some potential uses include the ability to search for clones directly or indirectly that are related to major gene differences, mutations, QTLs and for genes showing changes in gene expression during a developmental time-course or in a tissue basis [74,77–80]. In grain crops, time-course studies of seed development, gene expression during meiosis, and responses to specific environmental stimuli will identify expressed genes [13]. This will assign function to ESTs that will serve as potential candidates for mapped qualitative or quantitative loci affecting important traits. Characterization of the expression patterns of genes involved in genotype-by-environment interactions may eventually help unravel the complexities of the phenomena [77,81]. The success necessitates the application of genomics to the rapid validation of gene function and mode of action. As one example, the development of C-box binding factors (CBFs) for enhanced freezing and drought tolerance has been rapidly advanced because of the improved understanding generated by genomics technologies [68]. Harnessing the full potential of genomics-assisted wheat breeding will require a multidisciplinary approach and an integrated knowledge of the molecular and physiological processes influencing tolerance to drought.
8 Conclusions
Drought stress is one of the major limitations to wheat productivity. To develop crop plants with enhanced tolerance of drought stress, a basic understanding of physiological, biochemical and gene regulatory networks is essential. Various functional genomics tools have helped to advance our understanding of stress signal perception and transduction, and of the associated molecular regulatory network. These tools have revealed several stress-inducible genes and various transcription factors that regulate the drought-stress-inducible systems (Table 1).
Some examples of transgenic wheats reporting effects under drought environment
| Target | Gene | Effect | Reference |
| Mannitol | mtlD | In pot experiment after 30 days, drought transgenic line had greater dry weight, plant height and tiller number than control plants | [82] |
| DREB | DREB1A | After water was withheld for 10 days, there was less wilting in transgenic lines than control | [83] |
| LEA proteins | HVA1 | Higher root and shoot biomass than controls under drought stress and improved recovery after drought | [84] |
| Lipid transfer protein | TaLTP1 | Increasing by water stresses, such as by treatment at several PEG concentrations and NaCl, by hormone treatments | [85] |
| Membrane protein associated-protein | TaVAP | Increasing in response to mild drought stress in the flag leaf | [86] |
Meanwhile, new initiatives are being made in the area of plant biotechnology, especially in the area of functional genomic efforts by public and private sector research establishments. Genomes of cereal species with simpler genetic systems such as rice has been sequenced and maize sequence information will become available in the near future. Species with more complex genomes such as wheat can benefit by the information that will become available from the other species such as rice and maize by using candidate gene approaches. It is necessary to develop capacities to handle massive amounts of data that will become available from large-scale profiling experiments. For example, improving transformation efficiency as well as selection strategies for high throughput mutagenesis experiments will be two critical areas of research for the ultimate assignment of function to the numerous wheat genes that are being discovered using the new genomic tools.
In a word, the elucidation of genomic regions associated with the expression of traits involved in drought adaptation, the novel genes discovery or the determination of their expression patterns in response to drought stress will provide the basis of effective engineering strategies leading to enhanced wheat germplasm for specific agroecological niches. For any molecular assessment to be performed, it is paramount to firstly establish the plant adaptation strategy to overcome drought. Further, an account of stress-inducible regulatory genes that have been transferred into wheat plants to enhance stress tolerance is discussed as possible modes of integrating information gained from functional genomics into knowledge-based breeding programs.
Acknowledgements
Zhao Chang-Xing would like to acknowledge financial support from Doctoral Foundation of Qingdao Agricultural University (630523) and Project of Shandong Provincial Education Department (J06K57).