

1 Introduction
The great economic and nutritional value of the maize (Zea mays) kernel is mainly due to its high starch content, as it represents approximately 75% of the mature seed weight. However, the protein complement (ca. 10% of the mature seed weight), mainly found in the form of zeins (storage proteins) is essential for human and animal feeding. Yet the question remains of why the selection for higher starch level irremediably results in less protein content, as illustrated by the Illinois Long-Term Selection Experiment, which is spanning over more than 100 generations of classical breeding [1]. The endosperm, which is the main storage compartment of the maize grain, is therefore important both for the breeder because it is a major component of maize yield, and for the physiologist in that it provides the foundation for seedling germination and initial growth. Now the use of molecular markers and genomics may help understanding the mechanisms and regulations involved and open new avenues in genetic breeding.
Cereal endosperm development is well characterized from a physiological point of view. Three main key phases are described including a total of five successive phenomena (see [2] in this issue). The “lag phase” takes place from a few hours following pollination to 12 days after pollination (DAP) and consists in three stages: coenocytic, cellularization and differentiation resulting in rapid endosperm expansion. The subsequent period ranging from 12 to 40 DAP is dedicated to kernel filling. Biochemically, it consists in the conversion of imported sucrose and amino acids into starch and storage proteins. Most starch present in the endosperm is thus synthesized within a 28-day period. Afterwards, the maturation-desiccation phase occurs, leading to the mature kernel at 70 DAP. An analysis of numerous kernel mutants allowed characterizing the main biochemical steps in storage compound synthesis. However, among the approx. 300 genes involved in visible endosperm phenotypes [3,4] only few of them have yet been characterized at the molecular level [5]. Concerning the nutritional quality of seed proteins, the recessive opaque-2 (o2) mutant is the best characterized because the proportion of lysine and tryptophan (two essential amino acids for animals) is increased in mutant kernels. This effect is caused by a 50–70% decrease in zein content (these storage proteins are poor in these two essential amino acids) associated with a limited overproduction of an albumin-globulin fraction containing enriched amounts of these two amino acids [6]. In addition, the mutation was found to exert many pleiotropic effects, most probably because, as soon as in the early 1990s, the O2 gene was shown to encode a transcriptional activator of the basic leucine-zipper family possibly targeting several genes [7,8].
High-throughput genomics and post-genomics approaches are now providing new tools for a better understanding of the genetic and biochemical networks operating during kernel development. In maize, at least 5000 different genes could be expressed during development, excluding storage protein genes [9]. However, about 35% of them are orphan genes, whose functions remain enigmatic, possibly corresponding to endosperm specific genes [9], as also observed in wheat (Triticum spp.) [10]. Using a 1400-gene microarray, the o2 mutation was shown to affect the expression of numerous maize genes involved in development and metabolism [11]. Messenger RNAs are the primary products of gene expression, but their levels are often weakly correlated to corresponding protein levels [12]. A proteomic analysis in maize dealing with a comparison between the o2 mutant and corresponding wild type revealed extensive changes in accumulation of the storage proteins zeins [13] and of other proteins, including pyruvate-Pi-dikinase (PPDK) [14]. Proteomics has also been used to examine differential protein accumulation during development in dicot seeds [15] and in grain from rice (Oryza sativa) [16], barley (Hordeum vulgare) [17], wheat [18] and maize, within the EU-Zeastar program [19,20].
Although the metabolic pathways for the synthesis of starch and storage proteins during the accumulation period have been thoroughly analyzed in a number of studies, the developmental aspects, at both transcriptomic and proteomic levels, are much less documented and limited to a very few enzymes or proteins [21,22]. Moreover, the existence of a relationship between enzyme activity and accumulation in the corresponding mRNA and protein has only been examined for some key enzymes [23,24]. However, to our knowledge, no comparison of the developmental expression of the endosperm storage pathways has been made encompassing biochemistry to proteomics and transcriptomics. The present paper reviews such studies that were mostly performed in the context of the EU-Zeastar program together with previous studies focusing on the opaque-2 endosperm mutant. This combined biochemical, transcriptomic and proteomic analysis yields an overview of the regulation of carbohydrate and storage protein metabolisms in developing endosperm in relation to general kernel metabolism.
2 Maize endosperm development: an integrated view of mRNA expression, protein accumulation and storage metabolism
2.1 A comparison of transcriptome and proteome data from 10 to 21 DAP
Large-scale analyses of gene expression have been undertaken to obtain an integrated view of the genetic and biochemical networks operating during grain development. The mRNA expression and proteome patterns were analyzed in the context of the EU program Zeastar in the F2 maize inbred line [25]. The cDNA libraries were developed at three developmental stages: 1) lag phase (10 DAP), 2) beginning of storage (14 DAP), and 3) maximum storage product accumulation rate (21 DAP). Expressed sequence tags (ESTs) for 711, 757 and 384 relevant clones, respectively, were obtained, and functions were determined by homology with sequences stored in databases. During the time course, the proportion of sequences with no clear function decreased from 35 to 20%, while a large increase in storage protein sequences (i.e., from 5% to 38%) was observed. Other major identified functional categories were “Metabolism” (11–13%), “Transcription-RNA processing-protein synthesis” (13–20%), “Protein destination” (5–9%), “Cellular communication” (3–9%) and “Cell rescue-defense” (4%). In the “Metabolism” category, the respiratory-pathway transcripts represented the largest proportion (25–37%) and showed a shift in favor of glycolysis at 21 DAP. At this stage, the proportion of genes involved in amino acid metabolism increased to 17% whereas quite surprisingly those associated with starch metabolism decreased to 7%. A reference proteome map was established at 14 DAP [19], using 2-D PAGE followed by extensive identification of protein spots by LC-MS/MS. Among the 632 protein spots processed, 504 were identified by matching against databases. Identified proteins were not only cytoplasmic but also nuclear, mitochondrial or amyloplastic. Similarly to the transcript profiling expression, “Metabolic process enzymes”, “Protein destination”, “Protein synthesis” and “Cell rescue-defense-cell death-aging” were the most abundant functional categories. A good agreement was generally found between category rank in the 10 DAP transcriptome and the 14 DAP proteome, as expected because of possible lag phase between transcription and mature protein accumulation. The only notoriously missing categories in the proteome were kinases and proteins for RNA processing.
2.2 Starch synthesis: from transcripts to enzyme activities
A focus on the enzymes of carbohydrate metabolism was performed over a 10 to 40 DAP time course by quantifying 30 related transcripts by Q-RT PCR, 409 proteins (excluding storage proteins), the activities of eight key enzymes and nine metabolites in these biochemical pathways. Two main distinct patterns were observed. First, the invertases and their resulting products (hexoses) were maximum from 10 to 21 DAP, which is consistent with the need of hexose in early development [26]. Second, the starch pathway enzymes appeared from 14 DAP, the transcript anticipating protein or activity level but both paralleled starch accumulation suggesting that, in most cases, transcriptional control was responsible for the global regulation of starch biosynthesis.
2.3 PPDK and starch/protein balance: a proteomic analysis
The accumulation pattern of 409 proteins was further examined at seven developmental stages spanning all phases of grain development from 4 to 40 DAP. Hierarchical clustering analysis allowed four main developmental profiles to be recognized as presenting a maximum accumulation at 4–10 DAP, 10–14 DAP, 14–30 DAP and 30–40 DAP, respectively (cf. Fig. 4 in [20]). Protein composition in each cluster was physiologically meaningful and allowed raising a new hypothesis. Cluster 1 (maximum accumulation at 4–10 DAP), which is covering the lag-phase including cellularization, cell division and cell wall deposition, is characterized by maximum expression of actin, tubulin and cell organization proteins, of respiration metabolism (glycolysis and Krebs cycle) and of protection against reactive oxygen species. The large amount of proteins involved in proteolysis is consistent with an important protein turnover likely associated to the switch from growth and differentiation to storage. From cluster 2 to clusters 3 and 4, as development proceeds, the relative abundance of metabolism related enzymes increases from 37 to 47%, in agreement with endosperm storage filling but a fair amount of the “Protein destination” category (trafficking, chaperones, degradation) reached also a maximum proportion (ca. 30%) in the period. Proteins involved in protein folding (chaperones, BiP, PDI) had maximum accumulation between 14 and 21 DAP, consistent with the timing of storage protein accumulation. In contrast, the diversity in the “protein synthesis-RNA processing” category decreased.
A close examination of the “Metabolism” category (cf. Fig. 5 in [20]) reveals some interesting trends such as a large decrease in abundance, after 10 DAP, of Krebs-TCA cycle enzymes relative to the glycolytic enzymes. This observation was interpreted in the light of the recent demonstration that starch accumulation in the endosperm, i.e. after 12 DAP, proceeds under hypoxic conditions [27]. The lack of oxygen preventing the functioning of the Krebs cycle is likely to influence the expression of the corresponding enzymes. By contrast, ATP needed for starch synthesis from imported sucrose is produced without oxygen by the sucrose synthase-Ugpase-Agpase cycle and by the first steps of glycolysis (Fig. 1).

The expression of PPDK (pyruvate-Pi dikinase) from 20 DAP, may play a central role in the starch/amino acid balance through the liberation of PPi that reverses the Agpase reaction and in aromatic/alanine-aspartate amino acid balance through the PEP/pyruvate ratio.
Accumulation of other enzymes involved in metabolite production, namely amino acids, carbon skeletons and starch also increased in the two “late” profiles (clusters 3 and 4) but the most prominent change is the sudden onset, from 21 DAP, of the PPDK protein. A functioning of the PPDK in parallel to that of pyruvate kinase (PK) offers the interesting property of regulating the PEP/pyruvate ratio, two organic acids being at the entry point for aromatic and alanine-aspartate amino acid families, respectively. The reversibility of the reaction catalyzed by PPDK has also consequences on ATP and PPi levels since when PPDK is acting in the PEP-to-pyruvate direction it is doing the same as PK, except that one more ATP is produced by PPDK than by PK. In the reverse direction, PPDK produces one AMP and one PPi, which is potentially important for starch synthesis because PPi accumulation is pushing the reversible activity of Agpase toward ADP-glucose degradation rather than ADP-glucose synthesis, a mandatory step for starch synthesis (Fig. 1). Thus PPDK may play a central role in storage product composition, since PPi production will reduce starch accumulation in favor of amino acid synthesis, especially aspartate and aromatic acids. To fully validate this new hypothesis on the role of PPDK in the starch/protein balance, further experiments would be needed, as for instance, the direct measurement of PPi concentration and demonstration of a relationship between PEP/pyruvate concentration and the ratio between aromatic amino acids vs aspartate-derived amino acids. However, further supporting arguments are coming from a previous report showing that one of the two genes encoding PPDK is transcriptionally controlled by O2 [13]. The role O2 has to be closely examined in this context.
3 Impact of Opaque-2 on kernel development, metabolism and starch/protein balance
Opaque-2 mutation (o2) has been recognized in the mid 20th century as affecting not only kernel pericarp aspect but more importantly to provide high-lysine grains, thus increasing the nutritional value for monogastric animals. However, the mutation has a negative effect on grain yield and numerous efforts have been made to correct this drawback. The question is difficult since the o2 mutation entails numerous pleiotropic effects, at the plant but also the biochemical level in grain [28,29] but also in leaves [30] (Fig. 2). Several studies compared gene expression between wild-type and o2-mutant near-isogenic lines, at the protein level [13,14,31] as well as mRNA level [32,33]. A microarray analysis of 1400 maize genes revealed that the 60 genes up-regulated more than three times in the mutant are related to stress responses, molecular chaperones and protein turnover, but also to amino acid synthesis, at the notable exception of branched chain amino acids. Interestingly, among the 66 down-regulated genes, many genes are involved in carbon, carbohydrate metabolism and branched chain amino acids [11]. A study focusing on three enzymes of the aspartate pathway producing lysine, threonine and methionine found a significant reduction of only the first two enzyme activities (AK, aspartate kinase; HSDH, homoserine dehydrogenase) in the mutant [34] compared to wild type. Transcripts level of genes encoding enzymes of starch biosynthetic pathway were also altered in o2 mutant grain as compared to wild type [28]. The overall picture is a large epistatic and differential effect of O2 on carbohydrate and amino acid metabolisms.
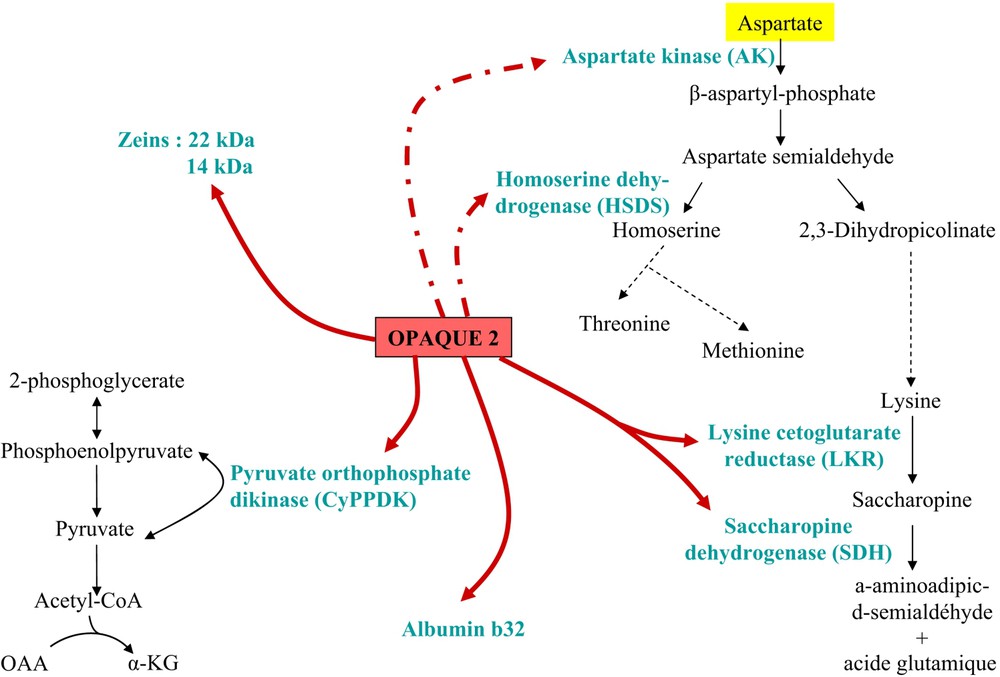
Regulatory effects of the Opaque-2 gene; known (solid arrows) and suspected (doted arrows) target genes of Opaque-2 protein; the enzymes encoded by the target genes are placed in a simplified metabolic pathway.
The pleiotropic effects encountered with mutants of the O2 gene may be explained by the fact that this gene encodes a transcriptional activator of the basic leucine-zipper family [7,8] expressed in the sub-aleurone layer from 10 DAP [35]. A direct effect on gene transcription was demonstrated on several zein and albumin classes [36,37] and on some other genes involved in various metabolisms. In particular, this transcription factor controls the transcription of the gene encoding the bifunctional enzyme LKR/SDH (lysine-ketoglutarate reductase/saccharopine dehydrogenase), which is the first enzyme of lysine catabolism. LKR/SDH activity, which temporally coincides with storage protein synthesis and accumulation, is strongly depressed in o2 mutants as compared to wild type [34,38]. This coordinated regulation suggests that potential excess of lysine not used for zein synthesis would be degraded in normal maize, although an explanation for this limitation in seed lysine accumulation is still lacking. In turn, this mechanism could act as a limiting factor for the synthesis of lysine-containing proteins, as can be deduced from the study of o2 mutants with increased lysine-containing protein content.
Another direct target of the O2 gene is the cyPpdk1 gene, which is down-regulated by the mutation [33]. This effect provides argument supporting our hypothesis for the role of PPDK in controlling the grain starch/protein balance, since O2 is reported to favor zein accumulation and to indirectly down-regulate starch metabolism enzymes. In addition, the o2 mutation also entails a decrease in the aromatic/other amino acids ratio, which may be explained by a depletion in PEP, the entry substrate of the aromatic amino acid pathway, due to a decreased PPDK expression [11]. This pivotal role of PPDK in the starch/protein balance is further supported by genetic data. In maize, PPDK is encoded by two genes located on chromosomes 6 and 8, respectively, but the cyPpdk1 gene is encoding the cytosolic isoform mainly expressed in cereal grains [39]. Its locus mapped at bin 6.05 where QTL (quantitative trait loci) for starch/protein ratio are co-located (http://www.maizegdb.org/). Furthermore, an association genetics analysis of the DNA polymorphism of cyPpdk1 and O2 genes in a representative panel of the maize line diversity (America and Europe) established both a significant association between cyPpdk1 gene polymorphism and starch/protein ratio and an epistatic interaction between cyPpdk1 and O2 genes for grain traits (Manicacci et al., in preparation).
4 Concluding remarks
The combined transcriptomic, proteomic and metabolic analyses of maize kernel development allowed a description of the shift in gene expression from the lag-phase to endosperm filling. Namely, the gene categories involved in cell division and expansion, but also in cell defence and metabolism were expressed in larger proportion in the first phase whereas carbohydrate and protein metabolism enzymes become predominant in the second phase although a high proportion of the “Protein destination” and “Protein synthesis” categories was also present. The dramatic increase of PPDK in the late filing period (21 DAP onwards) can be interpreted as providing a switch in the starch/protein balance because i) of the negative effect of PPi on the Agpase, a key-enzyme of starch synthesis, and ii) of the role of PEP in aromatic amino acid synthesis. The data on the Opaque-2 gene can explain the pleiotropic effects observed with o2 mutants as this transcription factor proved to affect lysine content through numerous enzymes of amino acid and carbohydrate metabolisms. The direct enhancing effect on PPDK transcription is likely critical in the competition between C and N metabolisms and supports the epistatic relationship between cyPpdk1 and O2 genes.
Acknowledgements
The financial support of the European Union (EU), 6th PCR, through the Zeastar project (QLK3-2000-00020), The Cell Factory section, is gratefully acknowledged.