

1 Introduction
The genetic code is degenerate. The relative frequency with which synonymous codons are used may vary widely from species to species, or from one gene (or a class of genes) to another. A large, and ongoing, body of literature deals with this subject under various headings, such as “codon usage”, “codon bias”, “codon adaptation”, etc. [1–17].
It is of significant interest to ask if, and to what extent, the genetic, molecular and cellular apparatus of each species is geared towards actual codon patterns of each sequence in the genome during translation and protein expression. The first part is obviously rhetorical, as there are plenty of indicators that such differences are not only present, but in fact crucial, and influence the steady state ratio of various proteins. Living organisms have very often quite biased preferences for some synonymous codons over other possible synonymous nucleotide triplets coding for the same amino acids. These differences and their variation have been extensively studied, however, no decisive governing rules have yet been discovered. Frequencies of codons for many species are in close correlation with their genome's GC contents, but the underlying forces governing this are not clear – it might be possible, that it is the GC content which is determining a genome's amino acids predilection for the specific codons being used and their bias. On the other hand it might be that reverse causative relationships are in operation: codons-specific amino acids usage is a driving factor for observed GC contents. Possible factors and forces driving synonymous codons usage postulated so far include, among many others: translational optimization [2–6], mRNA structural effects [7], protein composition [8], and protein structure [9], gene expression levels [2,10], the tRNA abundance differences between different genomes, and tRNA optimization [11–13], different mutation rates and patterns [14]. Also, some other possibilities were hypothesized, like local compositional bias [15], and even gene lengths might play a role too [16].
It is clear, that many interesting biological mechanisms underlie the basic phenomenon of genetic code degeneracy. One of its aspects, however, has not been studied until now, namely, the question dealing with the sequential order of occurrence of synonymous codons. To what extent this order is characteristic for a gene, to what extent for a set of genes, or a genome. Are there some rules governing such an order? How can one measure the order of synonymous codons, and compare different orders? Obviously, an order of elements in a linear set is a different property, than the frequency of elements in the set. The amino acid composition of a protein (which formally is exactly equivalent to the synonymous codon frequency, or codon usage, of a protein coding sequences) carries much less information than the amino acid sequence of such protein, which in turn is less information intensive than a corresponding nucleotide sequence coding the same protein.
This question can be formulated more precisely. Let us consider a given frequency of synonymous codon usage characteristic for a gene. There is a very large number of different orders in which the synonymous codons can appear sequentially along the gene without changing either the amino acid sequence of the encoded protein, or the codon usage of the gene. For all practical purposes this number is infinite, since the number of permutations of synonymous codons in a gene is comparable to the number of permutations of amino acids in a protein.
However, there are few means available experimentally to actually attempt even simplest direct measurements of the factors involved. Here, we describe an in silico method, which, as far as we know, is the first attempt to tackle the problem of the sequential order of synonymous codons. We call it ISSCOR (Intragenic, Stochastic Synonymous Codon Occurrence Replacement) for following reasons: (i) it analyses a original single gene, the nucleotide sequences of an original protein coding sequence (ORF). Of course, after analyzing several individual genes, the results can be computed for a set of genes, or a complete genome; (ii) it is essentially a Monte-Carlo approach. Synonymous codons, which occur at different positions of an ORF are replaced randomly, with the frequencies given by the codon usage of the whole genome. The method generates nucleotide sequences of non-original ORFs, which have identical codon usages, and would encode identical amino acid sequences. It is equivalent to random permutations of the synonymous codon sequence.
We have chosen to test the application of the ISSCOR method on the genome of Helicobacter pylori. This organism was selected for two reasons: (i) the H. pylori codon usage is not dominated by biased mutation patterns and displays a striking absence for transitionally mediated selection among synonymous codons [17]; and (ii) the H. pylori genome shows a striking periodic oscillations of it's nucleotide sequence, and it was hypothesized that these oscillations could be related to the usage of particular synonymous codons for constructing α-helical protein folds [18]. To this end we have retrieved (as of October 2006) from TIGR the complete set of H. pylori protein coding sequences, however, due to the needs of the ISSCOR algorithms, only those containing unambiguously assigned nucleotides (A, C, G, and T, in standard notation) have been used – this resulted in 1574 ORFs.
2 The concepts and methods used
2.1 The concept of Intragenic, Stochastic Synonymous Codon Occurrence Replacement
For the reasons outlined briefly above in the Introduction, we describe here the method of calculating the whole genome codon-pair pattern profiles, as well as assessing their significance. Previously [18–21] we have described alignment free approaches to the problem of comparison and analysis of complete genomes, and some techniques enabling us to cope with the sparseness of the n-gram type of genomic information representations. The problem of sparse occurrence matrices is not only present, but even more pronounced when dealing with the number of permutations of the possible synonymous codons. Calculating the set of n-grams for such occurrences will lead to a vector representation, which is severely sparse, especially for higher n-grams lengths, and hence to very poor statistics. To alleviate this problem, we propose here a hybrid approach. Namely, when computing counts of codon-pair patterns – separated by codon sub-sequences of differing length – the actual composition of these spacer sub-sequences will be neglected. However, when such partial counts are used as a composite set, poor statistics are no longer a hindering obstacle, and the complete information about particular n-gram frequencies profile is preserved, albeit in a distributed and convoluted form.
There are only two requirements fundamental for the ISSCOR method: (a) the amino acid sequence of every protein in the whole genome must be strictly preserved; (b) random shuffling of synonymous codons will be performed separately within each codon-degeneracy equivalence group using uniform probability distribution (thus, the codon usage profile for the whole genome, after the synonymous codon shuffling will be preserved within stochastic bounds of uniform probability distribution, and the overall codon usage of the whole genome will not be changed).
For every protein coding gene, with its original nucleotide sequence in a genome i we generate, by a Monte Carlo approach, a set of equivalent nucleotide strings () which have the following properties:
- • they have the same nucleotide lengths as the ;
- • they have the exactly the same amino acid sequence as the (i.e., the proteins translated from the are identical);
- • they have, on average, the same codon usage frequencies (i.e., the codon triplet occurrence frequency) as the whole genome, and if the gene under study has no codon bias, the same codon usage as ;
- • they have in the vast majority of cases a synonymous codon order different from the original sequence .
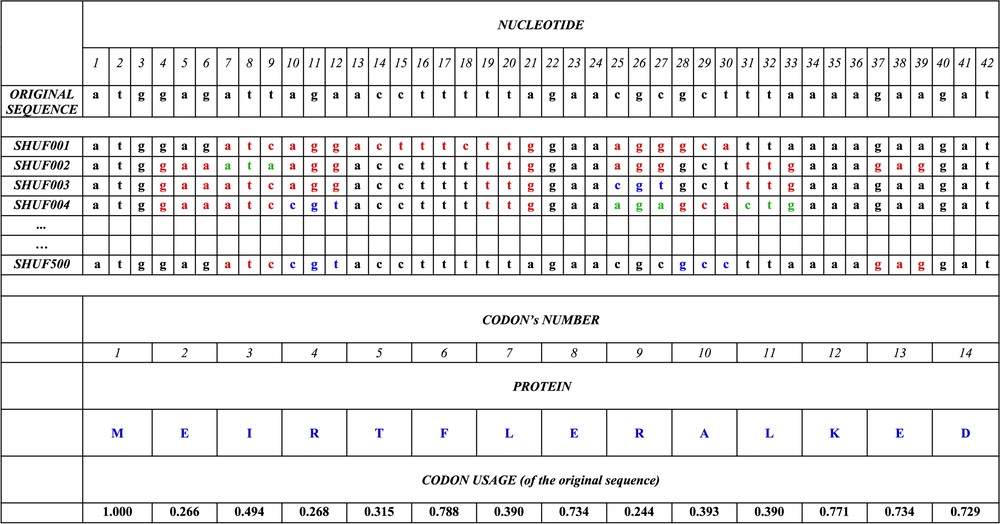
Example of synonymous codon replacements by the ISSCOR method, preserving the amino acid sequence of a gene. The beginning of (the first 14 codons) the original, genuine sequence of the gene HP1355 (coding for the nicotinate–nucleotide phosphorylase) is shown in the first line; the synonymous codons are replaced randomly using the frequencies given in the last line. The first run of replacement's (SHUF001) produces the nucleotide sequence, where synonymous codons, which are different from original are shown in red; in successive runs (SHUF002, 003, 004, until 500), other replacements appear (shown in green and blue). The frequency of occurrence of each of the 144 BiHex patterns (Table 3) is calculated for every run of replacements, i.e., separately for sequences SHUF001,002,…,500; finally the averages, their standard deviations; and deviates are calculated as explained in the text. The amino acid sequences are, of course, identical.
Therefore, the ISSCOR method allows comparing the original codon sequence with an ensemble of different synonymous sequences-coding for the same amino acids sequence.
2.2 The computational procedure
Mathematically speaking, first, the whole genome's codon usage frequencies are determined for each species i, and on that basis probabilities of replacement are calculated separately for each codon-degeneracy equivalence group d:
| (1) |
Then, successively for each codon in a gene the procedure of it's synonymous random replacement is performed based on probabilities according to Eq. (1). Finally, the resulting shuffled sequences are determined, and compared to the original sequence of the gene.
To this end we need, first of all to calculate series of codon-pair pattern1 occurrence matrices, designed henceforth as , for the original genome i. The method described here is applicable to any genome, and is independent of the respective genome sizes.
The method involves several steps (although, depending on the actual purpose at hand, not all the chain will be always necessary). For completeness sake, we present them here sequentially, to facilitate understanding.
First, for each protein coding sequence, we determine a complete matrix of all codon-pair patterns. Obviously, in a protein coding gene, there are at most 3904 () unique codon-pair patterns. For a given sequence V, and the all codon-spacer lengths, in order to calculate observed values of a particular codon-pair pattern (), first we need to construct a series of matrices (occurrence matrices). Each element of every matrix contains the counted sum of all specific codon-pair patterns (), separated by a string of other codons present in this sequence, where the λ denotes the number of other codons separating the given codon-pair pattern (). Using a sliding window of the length nucleotides, and starting at the position m, we would scan the whole sequence V, calculating elements of the matrix by the formula:
| (2) |
Comparisons involve matches between the predefined codon-pair patterns, of the first codon , taken together with the second codon .
That is, a particular positional comparison p involves only one nucleotide from the first codon , and one nucleotide from the second codon , ignoring all four remaining nucleotides, which corresponds to a BiHex pattern (see footnote 1). Thus, there are, e.g., nine BiHex patterns containing the adenine (A) at any position in a first codon, together with the cytosine (C) at any position in a second codon, etc. Obviously, when , one has an adjacent codon-pair pattern (hexanucleotide), for it is a nonanucleotide, and so on. Note, that since these are ordered counts, each starting at the sequence's 5′-terminus, the matrices are not symmetrical, that is the count of the hexon () is different from the count of the hexon (). For each of species i, their hexon counting is repeated for all sequences of the whole genome separately, but the results are then summed up for all sequences, and all respective particular pattern comparisons p. Therefore matrices should be considered as a raw, spacer λ dependent, representation of sui generis species specific occurrences of their hexons unique patterns.
To make the results independent of a particular genome size (or a subset of genes), we propose to calculate how much the number of actually observed hexons in the original genome, differs from the mean number of the corresponding hexons, observed after performing N random ISSCOR genome permutations, divided by the standard deviation observed in the shuffled genomes:
| (3) |
- is a deviate of the results for, e.g., the pattern , that is for the all codon combinations comprising the nucleotide A at the second position in the first codon, and the nucleotide T at the first position of the second codon – the border codons being separated by the number λ of other codons;
- are the numbers of the actually observed occurrences for any given hexon in the unperturbed, whole genome;
- are the numbers of occurrences for any given hexon pattern counted after codons of the whole genome have been shuffled randomly (as described by the above), thus the is a mean number of such occurrences after the N such random shuffles;
- is a standard deviation for all occurrences of a given hexon pattern, after N random shufflings of the whole genome.
We have verified that is sufficient to ensure satisfactory randomness since the results are practically identical for , 1000, and 1500.
3 Results and discussion
In order to facilitate the comprehension of the BiHex concept, and that of the first results obtained, we shall begin by an example, the detailed analysis of just one particular BiHex pattern: xxG-xxG for . It should be remembered that for all the examined λs there are 4455 () different and informative2 BiHex patterns, all of which have been computed in this work.
3.1 Detailed description of an example: analysis of just one BiHex pattern “xxG-xxG” for demonstrates that specific non-adjacent codon-pairs are strongly constrained in the original protein coding genes
This BiHex pattern, (where the symbol , or in a shorthand notation, denotes the BiHex of the pattern number 131, and with the codon-spacer ) can be written explicitly as xxGyyyyyyyyyyyyyyyyyyxxG. It corresponds to all the oligonucleotides (24 mers; as previously described, the “x” denotes any nucleotide found in the initial and terminal codons of a given BiHex pattern, and the “y” denotes any nucleotide constituting the spacer part between the respective initial and terminal codons of such a BiHex pattern), occurring in the reading frame zero, of the Helicobacter pylori protein coding genes. The analysis comprises several successive steps:
- (1) The number of occurrences of the xxGyyyyyyyyyyyyyyyyyyxxG pattern is counted in each of the 1574 original genes (in the example given in Fig. 1 this pattern is absent in the segment shown for the original sequence of the gene HP1355, but it is present in the shuffled sequences SHUFF001, SHUFF002, and SHUFF003, of this gene between the nucleotide positions 6 and 27, 12 and 33, and again 12 and 33, respectively). Altogether there are 21 819 occurrences of the BiHex pattern xxG-λ6-xxG in all the genes of Helicobacter (, Table 2A).
- (2) By the use of the ISSCOR method one generates 500 shuffled synonymous sequences for each gene ( nucleotide strings) and then calculates, as previously, the number of occurrences of the same BiHex pattern xxG-λ6-xxG which was found to be present, on average, 20 264 times in the ensemble of permuted sequences (, Table 2B).
- (3) The difference, , in the number of occurrences of this pattern between the observed original sequences, and the calculated average shuffled sequences is highly significant, whether measured by , or standard deviation , for one degree of freedom the probability is (Figs. 2A and 2B), and the deviate is more than 10 standard deviations (Fig. 2B). Therefore, some codon-pair hexons must be over-represented in the original genome.
- (4) Figs. 2A–2B and Tables 2A–2C prove this point. The BiHex pattern xxG-xxG is constituted by 240 different hexons (i.e., codon-pairs, 15 sense codons xxG upstream, and the same plus the TAG stop codon at the terminal position of an ORF, downstream). A priori, hexons can be allocated, by their number of occurrences in the genome, into three classes: (i) those which are significantly more abundant in the original genome than in the shuffled ones, (ii) those where the converse is true, and (iii) those where the difference between the original and the shuffled is too small to be of significance. It is apparent from Tables 2A–2C that the first class of hexons predominates: 29 different hexons occur more frequently in the original gene sequences than in the shuffled ones, and only one hexon (AAG-ACG) occurs less frequently. This result explains not only that the BiHex pattern xxG-λ=6-xxG is more abundant in the original, (observed) values than in the expected, (calculated from the shuffled) ones. It points also to the notion that the observed over-represented hexons exceed very strongly the expected values: the individual values, in the majority of cases, are greater than 7 (which corresponds to the E values ), and in several instances, e.g., GCG-λ=6-GCG, GTG-λ=6-GTG, GTG-λ=6-TTG, the values are greater than 11, for one degree of freedom, E values . Figs. 2A and 2B summarize the significance of individual hexons, and allows one to draw a few conclusions. Individual hexons are not distributed evenly, but rather in groups, which display some common characteristics. For instance, hexons containing CGG codon are never over-represented in their occurrence, and those containing ACG are very rarely over-represented. In contrast, hexons containing GGG or GTG codons are frequently over-represented. When the 240 different, individual hexons are divided into 16 simpler categories, by summing up the number of occurrences of all hexons containing a given codon, either at the 5′ or at the 3′ terminus of the respective polynucleotide chain, a very clear picture is obtained (Fig. 2B). Original gene sequences differ from the average permuted ones mainly by a higher frequency of codon-pairs containing either GGG or GTG, and less frequently by those containing TTG, GCG, GAG, while all the remaining hexons which contain other xxG codons, represent only a very minor contribution.
Detailed analysis. Observed number of occurrences of hexons (i.e. codon-pairs) in the original genome for the BiHex pattern xxG-λ=6-xxG.
| Observed | AAG | ACG | AGG | ATG | CAG | CCG | CGG | CTG | GAG | GCG | GGG | GTG | TAG | TCG | TGG | TTG | Sum |
| AAG | 250 | 65 | 109 | 172 | 61 | 28 | 15 | 41 | 224 | 168 | 236 | 292 | 10 | 34 | 65 | 294 | 2064 |
| ACG | 84 | 56 | 45 | 112 | 30 | 22 | 1 | 23 | 79 | 97 | 137 | 131 | 1 | 28 | 32 | 147 | 1025 |
| AGG | 93 | 40 | 61 | 83 | 20 | 17 | 3 | 24 | 86 | 83 | 78 | 127 | 2 | 29 | 38 | 122 | 906 |
| ATG | 189 | 96 | 98 | 254 | 76 | 35 | 10 | 50 | 207 | 226 | 224 | 321 | 9 | 46 | 77 | 358 | 2276 |
| CAG | 67 | 26 | 20 | 60 | 22 | 6 | 6 | 22 | 61 | 61 | 60 | 80 | 0 | 12 | 24 | 80 | 607 |
| CCG | 33 | 14 | 13 | 41 | 16 | 10 | 4 | 8 | 27 | 36 | 37 | 61 | 1 | 9 | 15 | 60 | 385 |
| CGG | 9 | 6 | 4 | 11 | 7 | 3 | 1 | 4 | 12 | 14 | 18 | 20 | 2 | 2 | 3 | 16 | 132 |
| CTG | 40 | 21 | 17 | 49 | 7 | 6 | 3 | 10 | 41 | 51 | 59 | 54 | 1 | 11 | 25 | 59 | 454 |
| GAG | 228 | 82 | 95 | 154 | 47 | 24 | 12 | 50 | 248 | 178 | 186 | 245 | 5 | 31 | 48 | 231 | 1864 |
| GCG | 201 | 78 | 99 | 218 | 56 | 31 | 15 | 42 | 178 | 275 | 267 | 326 | 7 | 41 | 73 | 331 | 2238 |
| GGG | 205 | 117 | 91 | 220 | 62 | 37 | 17 | 42 | 209 | 290 | 330 | 349 | 4 | 49 | 89 | 338 | 2449 |
| GTG | 298 | 136 | 102 | 310 | 66 | 71 | 12 | 48 | 239 | 326 | 367 | 477 | 9 | 68 | 124 | 444 | 3097 |
| TCG | 43 | 25 | 19 | 47 | 15 | 4 | 0 | 7 | 34 | 34 | 41 | 51 | 2 | 13 | 13 | 69 | 417 |
| TGG | 67 | 38 | 32 | 80 | 16 | 16 | 5 | 13 | 53 | 78 | 102 | 111 | 4 | 17 | 37 | 95 | 764 |
| TTG | 280 | 146 | 119 | 298 | 87 | 39 | 16 | 78 | 275 | 299 | 361 | 437 | 11 | 63 | 103 | 529 | 3141 |
| Sum | 2087 | 946 | 924 | 2109 | 588 | 349 | 120 | 462 | 1973 | 2216 | 2503 | 3082 | 68 | 453 | 766 | 3173 | 21 819 |
Detailed analysis. Calculated averages of occurrences of hexons (i.e. codon-pairs) in the shuffled genome for the BiHex pattern xxG-λ=6-xxG.
| Averages | AAG | ACG | AGG | ATG | CAG | CCG | CGG | CTG | GAG | GCG | GGG | GTG | TAG | TCG | TGG | TTG | Sum |
| AAG | 240 | 89 | 88 | 159 | 62 | 31 | 11 | 42 | 206 | 189 | 196 | 273 | 9 | 36 | 66 | 288 | 1985 |
| ACG | 94 | 57 | 37 | 102 | 28 | 17 | 5 | 21 | 82 | 106 | 105 | 121 | 3 | 19 | 35 | 143 | 972 |
| AGG | 84 | 37 | 42 | 84 | 23 | 13 | 5 | 17 | 77 | 80 | 91 | 114 | 3 | 16 | 34 | 117 | 837 |
| ATG | 200 | 103 | 93 | 254 | 59 | 33 | 12 | 54 | 191 | 221 | 240 | 312 | 5 | 45 | 77 | 369 | 2269 |
| CAG | 60 | 26 | 24 | 50 | 20 | 10 | 3 | 11 | 54 | 51 | 51 | 70 | 2 | 11 | 16 | 77 | 536 |
| CCG | 31 | 19 | 13 | 35 | 9 | 6 | 2 | 7 | 29 | 31 | 33 | 42 | 1 | 6 | 15 | 49 | 328 |
| CGG | 10 | 5 | 5 | 10 | 3 | 2 | 1 | 2 | 10 | 10 | 11 | 14 | 0 | 2 | 4 | 15 | 105 |
| CTG | 42 | 19 | 17 | 43 | 11 | 7 | 2 | 10 | 37 | 43 | 44 | 59 | 2 | 9 | 14 | 71 | 430 |
| GAG | 221 | 84 | 82 | 139 | 55 | 28 | 10 | 35 | 217 | 172 | 172 | 219 | 6 | 33 | 48 | 238 | 1759 |
| GCG | 191 | 97 | 84 | 222 | 53 | 34 | 11 | 45 | 171 | 224 | 234 | 276 | 6 | 39 | 68 | 305 | 2059 |
| GGG | 182 | 119 | 85 | 219 | 51 | 34 | 11 | 43 | 171 | 250 | 282 | 304 | 5 | 44 | 85 | 292 | 2177 |
| GTG | 256 | 130 | 110 | 272 | 67 | 49 | 14 | 55 | 222 | 285 | 331 | 409 | 9 | 54 | 107 | 378 | 2749 |
| TCG | 36 | 19 | 16 | 42 | 11 | 5 | 2 | 9 | 32 | 40 | 41 | 52 | 1 | 8 | 14 | 61 | 390 |
| TGG | 64 | 34 | 29 | 80 | 15 | 12 | 4 | 14 | 53 | 72 | 88 | 103 | 3 | 13 | 37 | 99 | 720 |
| TTG | 289 | 129 | 121 | 294 | 74 | 47 | 15 | 71 | 252 | 292 | 306 | 404 | 11 | 58 | 100 | 483 | 2947 |
| Sum | 2001 | 968 | 846 | 2005 | 539 | 328 | 107 | 435 | 1805 | 2068 | 2226 | 2773 | 67 | 392 | 721 | 2984 | 20 264 |
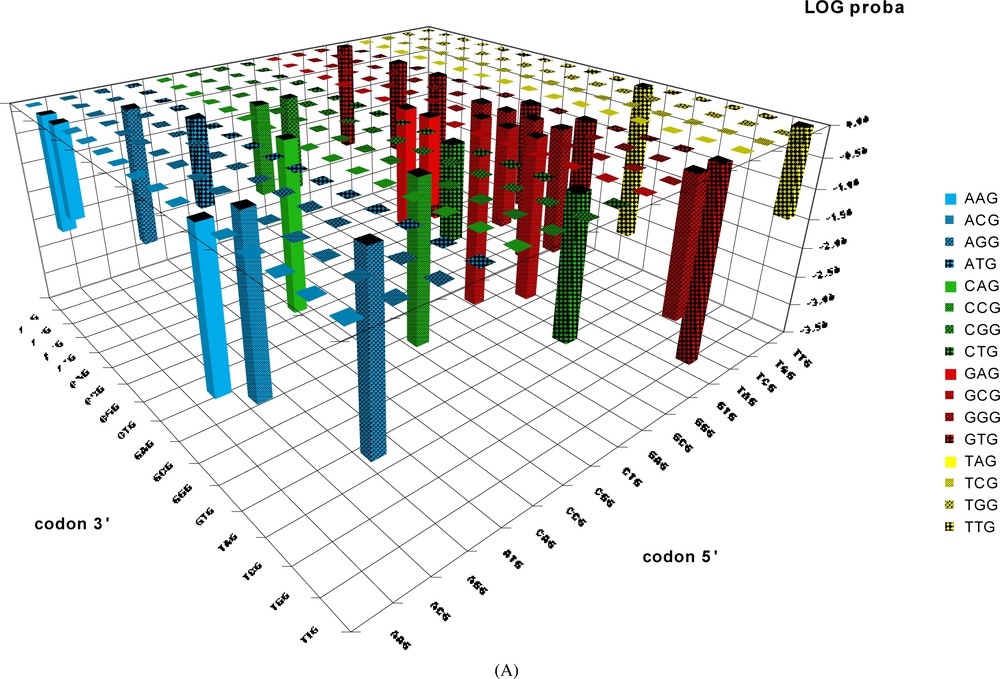
Probability of occurrence by chance of codon-pairs corresponding to the BiHex pattern xxG-λ=6-xxG. The probabilities (shown on a vertical axis in a log. scale) of occurrence by chance of the 240 hexon patterns were calculated from the values given by Table 2C (only the patterns giving probabilities <0.05 are shown). The patterns should be read in the order: codon 5′–codon 3′; thus the probability for the pattern TTGyyyyyyyyyyyyyyyyyyGCG is close to 10−3, and that of GTGyyyyyyyyyyyyyyyyyyTTG is close to 10−4, etc.
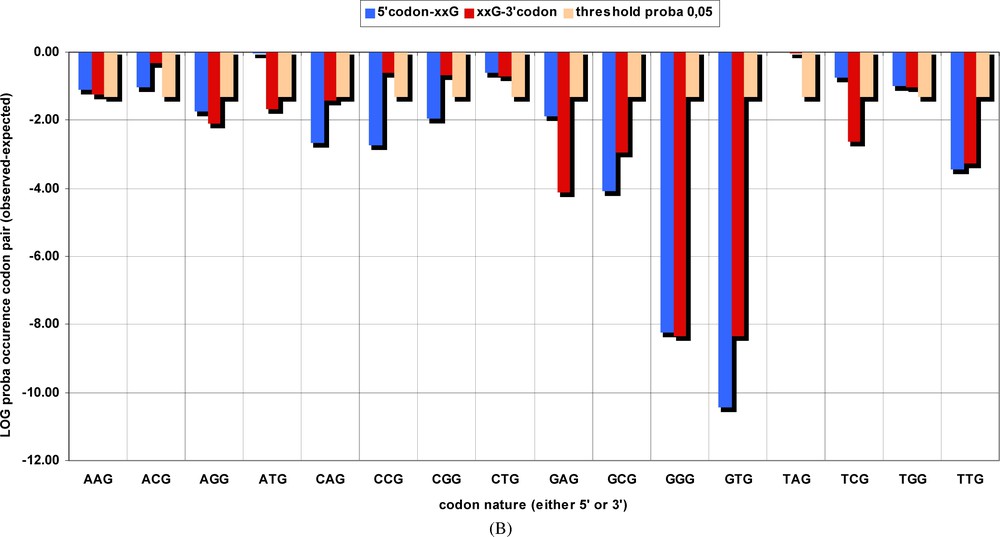
Probability of occurrence by chance of groups of codons pairs, corresponding to the BiHex pattern xxG-λ=6-xxG. The number of occurrences of all hexons beginning (in blue) with the codons given in the abscissa, or those terminated (in red) by them, were summed up for the original genes (Table 2A) and for the permuted ones (Table 2B). The and probabilities were calculated, and the latter are shown on the ordinate in a log scale. Thus, the probability for the GTG-λ=6-xxG pattern is close to 10−10, etc.
Detailed analysis. Yates corrected values for the difference between the Observed and the Calculated occurrences of hexons (i.e. codon-pairs) for the BiHex pattern xxG-λ=6-xxG.
| Yates Chi2 | AAG | ACG | AGG | ATG | CAG | CCG | CGG | CTG | GAG | GCG | GGG | GTG | TAG | TCG | TGG | TTG | Sum |
| AAG | 0.34 | 6.36 | 4.77 | 0.99 | 0.00 | 0.27 | 1.12 | 0.01 | 1.56 | 2.30 | 8.07 | 1.27 | 0.06 | 0.04 | 0.01 | 0.11 | 3.09 |
| ACG | 0.87 | 0.01 | 1.75 | 0.91 | 0.14 | 1.00 | 2.21 | 0.19 | 0.06 | 0.64 | 9.53 | 0.69 | nc | 3.75 | 0.12 | 0.10 | 2.84 |
| AGG | 0.97 | 0.11 | 7.85 | 0.00 | 0.19 | 1.19 | 0.70 | 2.65 | 0.91 | 0.10 | 1.65 | 1.33 | nc | 10.11 | 0.41 | 0.14 | 5.63 |
| ATG | 0.58 | 0.46 | 0.26 | 0.00 | 4.37 | 0.11 | 0.10 | 0.21 | 1.21 | 0.08 | 0.98 | 0.23 | nc | 0.00 | 0.00 | 0.30 | 0.02 |
| CAG | 0.67 | 0.00 | 0.63 | 1.93 | 0.09 | 1.00 | nc | 9.84 | 0.83 | 1.66 | 1.45 | 1.39 | nc | 0.06 | 3.22 | 0.08 | 9.33 |
| CCG | 0.06 | 0.98 | 0.00 | 0.75 | 4.62 | 1.64 | nc | 0.02 | 0.11 | 0.57 | 0.38 | 8.68 | nc | 0.73 | 0.00 | 2.37 | 9.68 |
| CGG | 0.08 | 0.17 | 0.14 | 0.00 | nc | nc | nc | nc | 0.32 | 1.31 | 3.21 | 1.83 | nc | nc | nc | 0.01 | 6.43 |
| CTG | 0.05 | 0.14 | 0.00 | 0.70 | 0.96 | 0.04 | nc | 0.00 | 0.30 | 1.48 | 4.43 | 0.33 | nc | 0.47 | 7.03 | 1.81 | 1.29 |
| GAG | 0.17 | 0.02 | 1.97 | 1.46 | 1.07 | 0.41 | 0.21 | 6.44 | 4.38 | 0.16 | 1.07 | 2.87 | 0.01 | 0.07 | 0.00 | 0.17 | 6.19 |
| GCG | 0.44 | 3.51 | 2.55 | 0.04 | 0.12 | 0.15 | 1.26 | 0.14 | 0.24 | 11.53 | 4.39 | 9.03 | 0.03 | 0.04 | 0.32 | 2.15 | 15.46 |
| GGG | 2.83 | 0.01 | 0.36 | 0.00 | 2.26 | 0.19 | 3.20 | 0.00 | 7.98 | 6.28 | 7.83 | 6.42 | 0.12 | 0.37 | 0.16 | 7.22 | 33.89 |
| GTG | 6.76 | 0.21 | 0.56 | 5.25 | 0.01 | 9.62 | 0.14 | 0.84 | 1.24 | 5.61 | 3.74 | 11.16 | 0.02 | 3.51 | 2.53 | 11.18 | 43.83 |
| TCG | 1.32 | 1.29 | 0.44 | 0.38 | 1.28 | 0.18 | 1.14 | nc | 0.05 | 0.85 | 0.00 | 0.01 | nc | 3.11 | 0.04 | 1.05 | 1.77 |
| TGG | 0.07 | 0.34 | 0.25 | 0.00 | 0.06 | 0.86 | 0.16 | nc | 0.00 | 0.36 | 2.20 | 0.50 | nc | 1.20 | 0.01 | 0.11 | 2.62 |
| TTG | 0.28 | 2.17 | 0.01 | 0.05 | 2.26 | 1.19 | 0.00 | 0.67 | 1.94 | 0.12 | 9.78 | 2.54 | 0.01 | 0.28 | 0.05 | 4.35 | 12.69 |
| Sum | 3.65 | 0.48 | 7.01 | 5.33 | 4.37 | 1.31 | 1.54 | 1.63 | 15.61 | 10.58 | 34.37 | 34.34 | 0.01 | 9.18 | 2.78 | 11.94 | 119.27 |
Contrary to numerous studies on the occurrence and the biases of adjacent codon-pairs [22–27], the analysis of non-adjacent codon-pairs is extremely limited. Only one study deals with this problem [23]. Based upon limited data available at that time (non-complete genome sequences of E. coli, S. cerevisiae, and H. sapiens) Hatfield and Gutman observed that the occurrence of non-adjacent codon-pairs is non random at long distances. Their data were obtained by pooling together all the possible 3904 codon-pairs into one bin only. The authors conclude that “the basis of this effect remains a mystery”. As far as we know, no analysis of this problem has been continued. Here we have shown that the different synonymous codon-pairs, specific for different distances, are responsible for the distant codon-pairs biases.
3.2 Perusal of all the informative 135 BiHex patterns for demonstrates, that all six positions of a pair of adjacent codons are constrained
It would be very tedious and undigestable to analyze in depth, in a manner analogous to that shown in Tables 2A–2C, all the 135 BiHex patterns listed in Table 1, for all the spacer codons λ from 0 to 32 (which would represent an equivalent of such individual tables!). Furthermore, this is not in the scope of the present article, which aims rather at the description of a novel method in computational biology of genomes. We believe that the introduction of the BiHex notation, both as a concept, and as a new method of computation, will be a successful shorthand for the description of profound and unexpected discoveries of the genuine, original order of synonymous codons whose nature and evolutionary significance remains to be understood. The results shown in Figs. 3A, 3B illustrate this notion.

BiHex patterns significantly deviating in the original genome for the adjacent codon-pairs (codon-spacer λ = 0). The number of occurrences of the BiHex patterns for λ = 0 (here, hexons correspond to hexanucleotides) are calculated for the original genome, and compared with their corresponding average number of occurrences in the 500 ISSCOR permutations (Eq. (3)). The difference, between original and the averaged permuted number of occurrences, is expressed in standard deviation units (STD). For clarity, we plot the deviates in two opposite directions – the patterns over-represented in the original above the zero line, and these under-represented below the zero line; with the negative sign. All BiHex patterns shown significantly deviate from randomness, some of them more than 100 STD (e.g., xxT-Txx – over-represented; and xxA-xxT – under-represented).
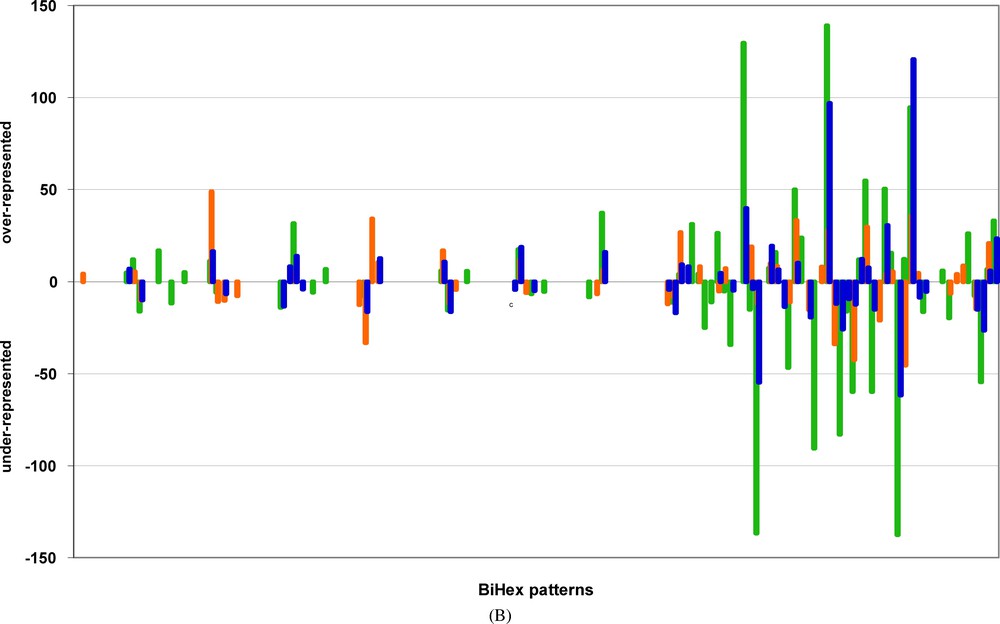
Examples of BiHex patterns significantly deviating in the original genome for the non-adjacent codon-pairs. The order of BiHex patterns, on the abscissa, is the same as in Fig. 3A, but their symbols are omitted for clarity, the numbering of patterns is given in Table 3. The ordinate shows the values calculated for the difference between the observed occurrences, and the calculated occurrence averages after 500 ISSCOR permutations. Only the significant averages (probabilities <0.05) are shown. For clarity, the values are plotted in opposite directions – the patterns over-represented in the original are above the zero line, and the under-represented below the zero line. The values corresponding to the codon-spacers: λ = 3 are green, for λ = 5 are orange, and for λ = 6 are blue. Notice, that value of 10 corresponds to the probability of 10−3, that of 20 to 10−6, that of 50 to 10−12, and that of 140 to 10−32.
A majority of BiHex patterns (109/135) significantly deviate in the adjacent codon-pairs, i.e., (Figs. 3A, 3B). Both over-represented and under-represented BiHex patterns are present in comparable numbers and with comparable amplitude of their deviates (compare, e.g., the BiHex pattern xxT-Txx, which is over-represented by more than 100 standard deviations (STD), with the pattern xxA-xxT, which is under-represented by more than 100 STD in the original gene sequences. The E values for such amplitudes are smaller than 100−100. It should be remembered, from the complete analysis of the BiHex pattern xxG-xxG (vide supra), that if a given BiHex pattern is strongly deviating, then numerous codon-pairs corresponding to such a pattern must be strongly deviating too, all in the same direction (i.e., all in “plus”, or in all “minus”). Therefore, one can conclude, that hexanucleotides xxTTxx (and also xxGCxx, xxAGxx, xxCAxx, etc.; Figs. 3A, 3B), in the reading frame 0, are strongly over-represented, while at the same time the hexanucleotides xxTCxx, xxGTxx, and xxTAxx, are under-represented in the H. pylori protein coding genes. It is well known that the fourth position [11,12] of a pair of adjacent codons is strongly constraint because of the properties of the translation machinery, and the interactions between the ribosomal A and P sites. It is therefore not surprising that the above-observed deviations, which all deal with the fourth position, are detected. The ISSCOR method and the BiHex concept simply allow to sieve out rapidly the nature of nucleotides present at the fourth position, pinpointing the nature of the nucleotides upstream, at the 1st, 2nd or 3rd position of hexanucleotides in the reading frame zero, and to estimate the statistical significance of the observed associations.
Much richer in unexpected results are the comparisons concerning the 5th and the 6th position of adjacent codon-pairs. In several particular patterns the 5th position of an, in frame, hexanucleotide is vastly over-represented (e.g., xxTxTx, xxCxCx, CxxxTx, and xxGxGx), while in others (e.g., xxGxAx, xxCxGx, and CxxxGx) it is strongly under-represented. In the 6th position, again numerous highly significant deviations are observed. The xxAxxT hexanucleotide is the most strongly deviating one amongst all analyzed. Its occurrence in the Helicobacter genes is the least frequent. Just the opposite is true for the hexanucleotides xAxxxC, xxAxxC, and xxGxxT, which are amongst the most frequently occurring; and the most significant patterns. There is no point here, however, in enumerating such a simplified catalogue. What our results demonstrate, is that all positions in a pair of adjacent codons are important, which means that the order of synonymous codons is an intrinsic characteristics of a genuine genome, and that this order can not be too dissimilar between different individual genes. Otherwise, the global occurrences of BiHex patterns would have been scrambled, and no significant deviations could have been uncovered in the complete set of genomic sequences. It is important to repeat once more, that the random permutations of synonymous codons by the ISSCOR method allow us to disentangle the significant deviations from the basal noise. They change neither the codon usage frequencies, nor the amino acid or protein sequences. They earmark and measure “pure” properties of the sequential order, and not the frequencies of the constituents (codons, amino acids) of that order.
In a comprehensive survey of codon-pair biases across ORFs from 16 genomes, Buchan et al. ([12], and references therein), have concluded that tRNA properties help shaping adjacent codon-pair preferences. Our results are compatible with this interpretation. They strengthen, however, the idea that the basis of selection of codon-pairs depends not only on the tetranucleotide combinations, but on the complete hexanucleotide combinations (with its first and last positions as well). Interestingly, our results were obtained with Helicobacter pylori, which is an organism amongst the least affected by the tRNA-based translational selection of synonymous codons [12,17].
3.3 Perusal of BiHex patterns at long range distances demonstrates that the sequential order of synonymous codons is constrained in a specific manner all along protein coding genes
Fig. 3A gives examples and demonstrates the “pure” order aspects at long-range distances along the genes. Highly significant deviations can be observed and measured for non-adjacent codon-pairs separated by 3, 5, or 6 codon spacers λ. Although less striking in their amplitude of differences than those observed for the adjacent codon-pairs (i.e., ), their significance leaves no doubt. Several BiHex patterns from the original genome differ from those of the permuted ones from 5 up to 15 STD units. These differences occur as well in the positive direction (over abundance), as in the negative direction (under abundance). These deviations correspond to E values smaller than 10−10. Importantly, for each set of distances along the genes a different set of significant BiHex patterns is detected. One can see, in the examples shown in Fig. 3A, that the BiHex patterns deviating at (orange) frequently do not coincide with those deviating at (blue). The property of “sui generis” constraints is therefore always true at long range, but the nature of specific codon-pairs, which are constrained at long distances is different. Just one example, the comparison of a 21-mer () oligonucleotide, and a 24-mer () oligonucleotide, illustrates this notion. We have verified, by the in depth analysis of the BiHex pattern xxG-xxG (vide supra), that this is true for the 240 codon-pairs, which constitute this pattern. These codon-pairs, which are conspicuous in the 24-mers, and significantly over-represented (Table 3), are not necessarily those, which are conspicuous and over-represented in 21-mers (data not shown), and vice versa.
In conclusion the remarkable and singular properties of polynucleotide chains described in this work are properties of the sequential order of synonymous codons within a gene, which must be under selective pressure. This sequential order is thus superimposed on the classical codon usage frequencies. This new dimension can be measured by the ISSCOR method, which is simple, robust, and should be useful for comparative and functional genomics.
Acknowledgements
J.P.R. was partially supported by the EU project SSPE-CT-2006-44405, also J.P.R. and P.P.S. were partially supported from the 352/6.PR-UE/2007/7 grant. We would like to thank Dr. C.J. Herbert for looking over the English. This contribution was presented for the first time at the international symposium “The Logic of Gene Regulation Networks” in the honor of Prof. René Thomas, Brussels, May 30–31, 2008.
1 As the repetitive use of the sub-phrase: “codon-pair pattern” may lead to possible confusion, and it might be tedious for the reader, we introduce here the term of hexon. Thus, it is important to give, and to distinguish several definitions of some frequently used notions:
- Codon-pair: two consecutive codons in the reading frame zero, located on the same strand, and within the same ORF (in the 5′–3′ orientation, i.e., Watson strand); there are such different codon-pairs, and different codon-pairs in an ORF terminated by a stop codon.
- Hexon: a codon-pair separated by an arbitrary number of other codons (λ); for the hexon is an adjacent codon-pair, corresponding to a hexanucleotide while for all other values of λ, the hexon corresponds to non-adjacent codon-pairs; for the hexon corresponds to a nonanucleotide, for it corresponds to a dodecanucleotide, etc. (for which only the first and the last codons are specified); in this work we shall explore hexons from to ; i.e., from hexanucleotides, to 102-oligonucleotides, for which there are different hexon patterns.
- Bigram: is a group of two – out of four possible – symbols (in the context of this work they are A, C, G, and T, clearly corresponding to the four DNA nucleotides).
- BiHex: is a bigram derived from a hexon; since each codon comprises three nucleotides, in the respective positions 1, 2, 3, and the analysis involves all possible positional combinations of the four nucleotides; at six positions, there are altogether 144 BiHex patterns () for each given value of λ, and for all values of λ explored here. Table 1 summarizes all the possible BiHex patterns.
| A | C | G | T | |
| A | Axx-Axx xAx-Axx xxA-Axx | Cxx-Axx xCx-Axx xxC-Axx | Gxx-Axx xGx-Axx xxG-Axx | Txx-Axx xTx-Axx xxT-Axx |
| Axx-xAx xAx-xAx xxA-xAx | Cxx-xAx xCx-xAx xxC-xAx | Gxx-xAx xGx-xAx xxG-xAx | Txx-xAx xTx-xAx xxT-xAx | |
| Axx-xxA xAx-xxA xxA-xxA | Cxx-xxA xCx-xxA xxC-xxA | Gxx-xxAx Gx-xxA xxG-xxA | Txx-xxA xTx-xxA xxT-xxA | |
| C | Axx-Cxx xAx-Cxx xxA-Cxx | Cxx-Cxx xCx-Cxx xxC-Cxx | Gxx-Cxx xGx-Cxx xxG-Cxx | Txx-Cxx xTx-Cxx xxT-Cxx |
| Axx-xCx xAx-xCx xxA-xCx | Cxx-xCx xCx-xCx xxC-xCx | Gxx-xCx xGx-xCx xxG-xCx | Txx-xCx xTx-xCx xxT-xCx | |
| Axx-xxC xAx-xxC xxA-xxC | Cxx-xxC xCx-xxC xxC-xxC | Gxx-xxC xGx-xxC xxG-xxC | Txx-xxC xTx-xxC xxT-xxC | |
| G | Axx-Gxx xAx-Gxx xxA-Gxx | Cxx-Gxx xCx-Gxx xxC-Gxx | Gxx-Gxx xGx-Gxx xxG-Gxx | Txx-Gxx xTx-Gxx xxT-Gxx |
| Axx-xGx xAx-xGx xxA-xGx | Cxx-xGx xCx-xGx xxC-xGx | Gxx-xGx xGx-xGx xxG-xGx | Txx-xGx xTx-xGx xxT-xGx | |
| Axx-xxG xAx-xxG xxA-xxG | Cxx-xxG xCx-xxG xxC-xxG | Gxx-xxG xGx-xxG xxG-xxG | Txx-xxG xTx-xxG xxT-xxG | |
| T | Axx-Txx xAx-Txx xxA-Txx | Cxx-Txx xCx-Txx xxC-Txx | Gxx-Txx xGx-Txx xxG-Txx | Txx-Txx xTx-Txx xxT-Txx |
| Axx-xTx xAx-xTx xxA-xTx | Cxx-xTx xCx-xTx xxC-xTx | Gxx-xTx xGx-xTx xxG-xTx | Txx-xTx xTx-xTx xxT-xTx | |
| Axx-xxT xAx-xxT xxA-xxT | Cxx-xxT xCx-xxT xxC-xxT | Gxx-xxT xGx-xxT xxG-xxT | Txx-xxT xTx-xxT xxT-xxT |
2 , see explanation in the legend of Table 3. There are therefore nine non-informative BiHex patterns.
| No. | BiHex | 5′ | 3′ | No. | BiHex | 5′ | 3′ | No. | BiHex | 5′ | 3′ | No. | BiHex | 5′ | 3′ | No. | BiHex | 5′ | 3′ | No. | BiHex | 5′ | 3′ |
| 1 | Axx-Axx | R;S | R;S | 25 | Gxx-Axx | – | R;S | 49 | xAx-Axx | – | R;S | 73 | xGx-Axx | S | R;S | 97 | xxA-Axx | aa_12 | R;S | 121 | xxG-Axx | aa_11 | R;S |
| 2 | Axx-Cxx | R;S | L;R | 26 | Gxx-Cxx | – | L;R | 50 | xAx-Cxx | – | L;R | 74 | xGx-Cxx | S | L;R | 98 | xxA-Cxx | aa_12 | L;R | 122 | xxG-Cxx | aa_11 | L;R |
| 3 | Axx-Gxx | R;S | – | 27 | Gxx-Gxx | – | – | 51 | xAx-Gxx | – | – | 75 | xGx-Gxx | S | – | 99 | xxA-Gxx | aa_12 | – | 123 | xxG-Gxx | aa_11 | – |
| 4 | Axx-Txx | R;S | L;S | 28 | Gxx-Txx | – | L;S | 52 | xAx-Txx | – | L;S | 76 | xGx-Txx | S | L;S | 100 | xxA-Txx | aa_12 | L;S | 124 | xxG-Txx | aa_11 | L;S |
| 5 | Axx-xAx | R;S | – | 29 | Gxx-xAx | – | – | 53 | xAx-xAx | – | – | 77 | xGx-xAx | S | – | 101 | xxA-xAx | aa_12 | – | 125 | xxG-xAx | aa_11 | – |
| 6 | Axx-xCx | R;S | S | 30 | Gxx-xCx | – | S | 54 | xAx-xCx | – | S | 78 | xGx-xCx | S | S | 102 | xxA-xCx | aa_12 | S | 126 | xxG-xCx | aa_11 | S |
| 7 | Axx-xGx | R;S | S | 31 | Gxx-xGx | – | S | 55 | xAx-xGx | – | S | 79 | xGx-xGx | S | S | 103 | xxA-xGx | aa_12 | S | 127 | xxG-xGx | aa_11 | S |
| 8 | Axx-xTx | R;S | – | 32 | Gxx-xTx | – | – | 56 | xAx-xTx | – | – | 80 | xGx-xTx | S | – | 104 | xxA-xTx | aa_12 | – | 128 | xxG-xTx | aa_11 | – |
| 9 | Axx-xxA | R;S | aa_12 | 33 | Gxx-xxA | – | aa_12 | 57 | xAx-xxA | – | aa_12 | 81 | xGx-xxA | S | aa_12 | 105 | xxA-xxA | aa_12 | aa_12 | 129 | xxG-xxA | aa_11 | aa_12 |
| 10 | Axx-xxC | R;S | aa_15 | 34 | Gxx-xxC | – | aa_15 | 58 | xAx-xxC | – | aa_15 | 82 | xGx-xxC | S | aa_15 | 106 | xxA-xxC | aa_12 | aa_15 | 130 | xxG-xxC | aa_11 | aa_15 |
| 11 | Axx-xxG | R;S | aa_11 | 35 | Gxx-xxG | – | aa_11 | 59 | xAx-xxG | – | aa_11 | 83 | xGx-xxG | S | aa_11 | 107 | xxA-xxG | aa_12 | aa_11 | 131 | xxG-xxG | aa_11 | aa_11 |
| 12 | Axx-xxT | R;S | aa_15 | 36 | Gxx-xxT | – | aa_15 | 60 | xAx-xxT | – | aa_15 | 84 | xGx-xxT | S | aa_15 | 108 | xxA-xxT | aa_12 | aa_15 | 132 | xxG-xxT | aa_11 | aa_15 |
| 13 | Cxx-Axx | L;R | R;S | 37 | Txx-Axx | L;S | R;S | 61 | xCx-Axx | S | R;S | 85 | xTx-Axx | – | R;S | 109 | xxC-Axx | aa_15 | R;S | 133 | xxT-Axx | aa_15 | R;S |
| 14 | Cxx-Cxx | L;R | L;R | 38 | Txx-Cxx | L;S | L;R | 62 | xCx-Cxx | S | L;R | 86 | xTx-Cxx | – | L;R | 110 | xxC-Cxx | aa_15 | L;R | 134 | xxT-Cxx | aa_15 | L;R |
| 15 | Cxx-Gxx | L;R | – | 39 | Txx-Gxx | L;S | – | 63 | xCx-Gxx | S | – | 87 | xTx-Gxx | – | – | 111 | xxC-Gxx | aa_15 | – | 135 | xxT-Gxx | aa_15 | – |
| 16 | Cxx-Txx | L;R | L;S | 40 | Txx-Txx | L;S | L;S | 64 | xCx-Txx | S | L;S | 88 | xTx-Txx | – | L;S | 112 | xxC-Txx | aa_15 | L;S | 136 | xxT-Txx | aa_15 | L;S |
| 17 | Cxx-xAx | L;R | – | 41 | Txx-xAx | L;S | – | 65 | xCx-xAx | S | – | 89 | xTx-xAx | – | – | 113 | xxC-xAx | aa_15 | – | 137 | xxT-xAx | aa_15 | – |
| 18 | Cxx-xCx | L;R | S | 42 | Txx-xCx | L;S | S | 66 | xCx-xCx | S | S | 90 | xTx-xCx | – | S | 114 | xxC-xCx | aa_15 | S | 138 | xxT-xCx | aa_15 | S |
| 19 | Cxx-xGx | L;R | S | 43 | Txx-xGx | L;S | S | 67 | xCx-xGx | S | S | 91 | xTx-xGx | – | S | 115 | xxC-xGx | aa_15 | S | 139 | xxT-xGx | aa_15 | S |
| 20 | Cxx-xTx | L;R | – | 44 | Txx-xTx | L;S | – | 68 | xCx-xTx | S | – | 92 | xTx-xTx | – | – | 116 | xxC-xTx | aa_15 | – | 140 | xxT-xTx | aa_15 | – |
| 21 | Cxx-xxA | L;R | aa_12 | 45 | Txx-xxA | L;S | aa_12 | 69 | xCx-xxA | S | aa_12 | 93 | xTx-xxA | – | aa_12 | 117 | xxC-xxA | aa_15 | aa_12 | 141 | xxT-xxA | aa_15 | aa_12 |
| 22 | Cxx-xxC | L;R | aa_15 | 46 | Txx-xxC | L;S | aa_15 | 70 | xCx-xxC | S | aa_15 | 94 | xTx-xxC | – | aa_15 | 118 | xxC-xxC | aa_15 | aa_15 | 142 | xxT-xxC | aa_15 | aa_15 |
| 23 | Cxx-xxG | L;R | aa_11 | 47 | Txx-xxG | L;S | aa_11 | 71 | xCx-xxG | S | aa_11 | 95 | xTx-xxG | – | aa_11 | 119 | xxC-xxG | aa_15 | aa_11 | 143 | xxT-xxG | aa_15 | aa_11 |
| 24 | Cxx-xxT | L;R | aa_15 | 48 | Txx-xxT | L;S | aa_15 | 72 | xCx-xxT | S | aa_15 | 96 | xTx-xxT | – | aa_15 | 120 | xxC-xxT | aa_15 | aa_15 | 144 | xxT-xxT | aa_15 | aa_15 |