

1 Introduction
The reassignment of the CUG codon from Leu to Ser occurs in at least 75 Candida species and in Pichia stipitis, Debaryomyces hansenii and Lodderomyces elongisporus [1,2], which form the so-callled CTG clade [3]. C. zeylanoides, C. dubliniensis, C. tropicalis, C. guilliermondii, C. albicans and many others translate CUGs ambiguously (Ser + Leu) and their proteins contain ∼97% of Ser and ∼3% of Leu at CUG positions, while C. cylindracea decodes CUGs as Ser only [4,5]. The change of identity of the CUG codon is mediated by a novel tRNACAGSer (Fig. 1), whose peculiar double recognition by SerRS and LeuRS leads to synthesis of two different aminoacyl-tRNAs, namely a Ser-tRNACAGSer and a Leu-tRNACAGSer, which compete for CUGs at the ribosome A-site during mRNA translation [6]. The mischarged leu-tRNACAGSer is neither edited by the LeuRS nor discriminated by the translation elongation factor 1 (eEF1A) and, consequently, Leu and Ser are incorporated into the proteome at CUG positions in vivo [7]. Apart from this, the tRNACAGSer contains a unique guanosine at position 33 (G33), which is located adjacent to the 5’ base of the 5’-CAG-3’ anticodon (Fig. 1), a position occupied by a critical uridine (U33) required for the U-turn of the anticodon-loop [8]. G33 induces a long-range distortion of the top of the anticodon stem of the tRNACAGSer and lowers its leucylation and decoding efficiencies [9,10]. Finally, the discriminator base (N73) of the tRNACAGSer is guanosine (G73) which is an identity element of Ser tRNAs (Fig. 1). This raises the intriguing question of how the LeuRS recognizes this tRNA because a single A73 to G73 mutation in S. cerevisiae tRNALeu converts its identity from Leu to Ser [11]. In other words, the C. albicans LeuRS should not recognize the tRNACAGSer as the tRNA identity elements are conserved between these two yeasts. But the tRNACAGSer contains A35 and m1G37 in its anticodon-loop, which are directly recognized by the LeuRS (Fig. 2) [11], suggesting that these two identity elements are sufficient for proper recognition of the tRNACAGSer by the LeuRS. This question needs to be clarified at the structural and enzymatic levels using X-ray crystallography of the LeuRS-tRNACAGSer complex and aminoacylation assays.
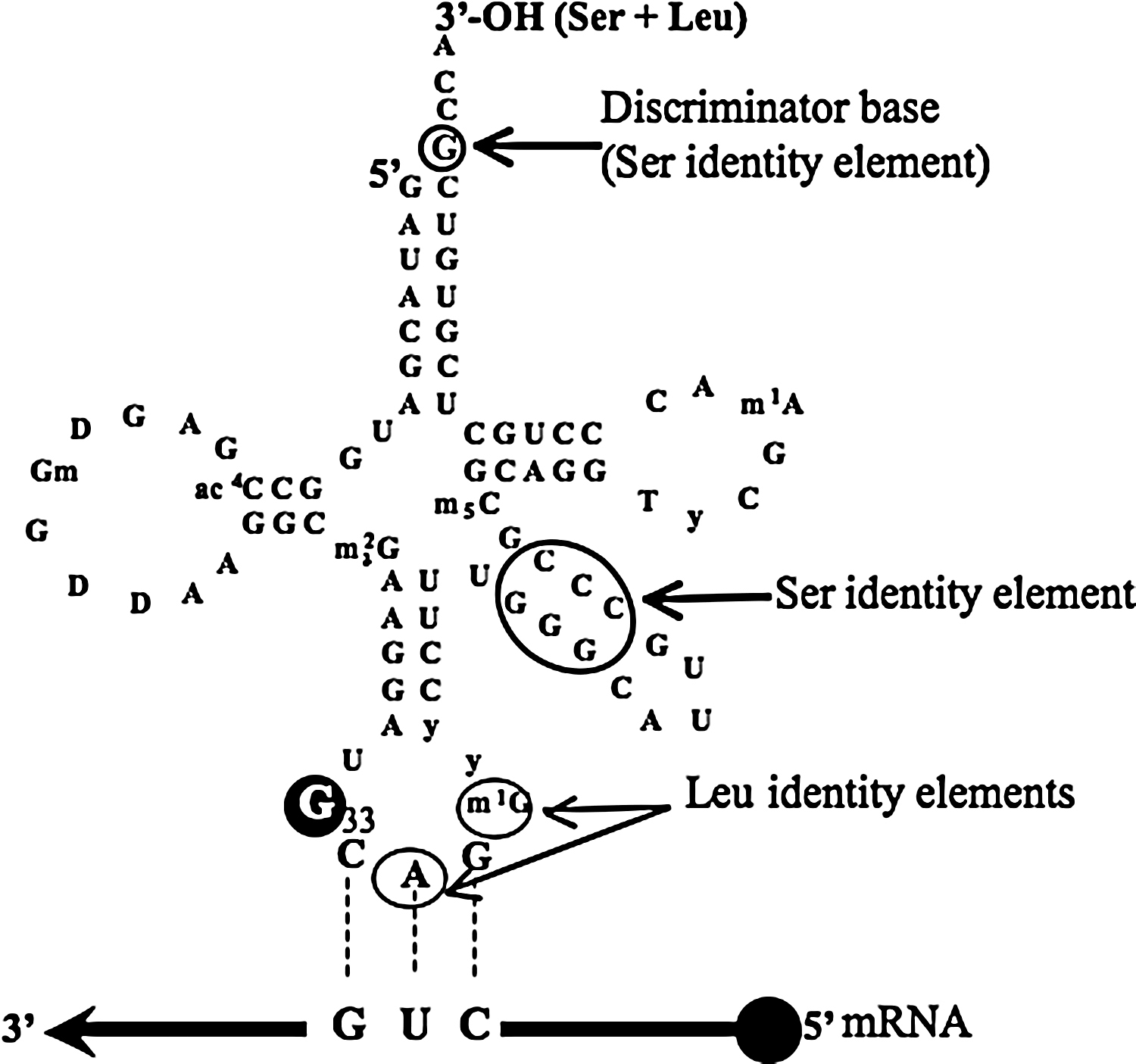
C. albicans tRNACAGSer. The C. albicans tRNACAGSer is an hybrid tRNA with identity elements for both leucyl and seryl tRNA synthetases (LeuRS and SerRS). Its anticodon arm is characteristic of leucine tRNAs and contains two leucylation identity elements, namely A35 and m1G37, whereas the acceptor stem and the variable arm are characteristic of serine tRNAs. The discriminator base (G73) and the G3C3 run of the extra-arm are characteristic of serine tRNAs [25].
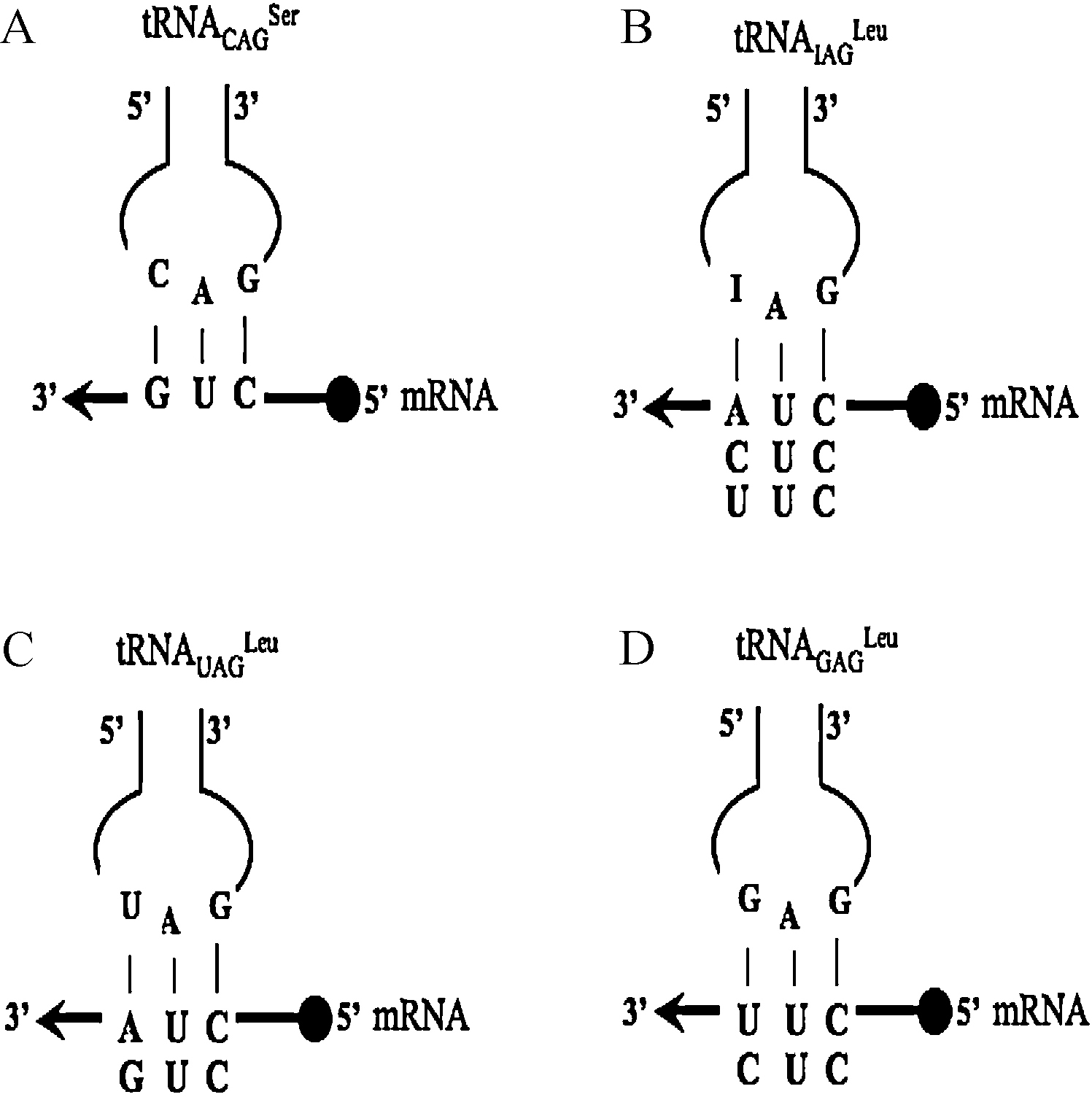
Decoding of the CUN codon family in C. albicans and S. cerevisiae. A, B. (In C. albicans CUG codons are decoded by a mutant serine tRNACAGSer while the CUU, CUC and CUA codons are decoded by a tRNAIAGLeu). C, D. In S. cerevisiae, a tRNAUAGLeu decodes CUA and CUG codons and a tRNAGAGLeu decodes the CUU and CUC codons.
2 The reassignment of CUG codons in the fungal CTG clade
The reassignment of CUGs in the fungal CTG clade strongly supports the “Ambiguous Intermediate Theory” of the genetic code, which postulates that codons are reassigned through ambiguous decoding [12–14]. Interestingly, such codon ambiguity erased 98% of the CUGs of the CTG clade ancestor through mutation to the frequently used Leu UUA and UUG codons. The “new” CUG codons present in extant species of the CTG clade evolved recently from codons coding for Ser or amino acids with similar chemical properties thus showing that the CUGs of the CTG clade species are phylogenetically unrelated to the CUGs of the other fungal species [3,15]. Comparative genomics and molecular phylogeny studies showed that the tRNACAGSer appeared 272 ± 25 million years ago, prior to the divergence between the Saccharomyces and Candida genera (170 ± 27 million years ago), via insertion of an adenosine in the anticodon of a serine tRNACGASer gene [15]. However, the evolutionary pathway of CUG reassignment has not yet been reconstructed and one does not fully understand how CUGs changed their identity. In particular, selective advantages produced by CUG ambiguity, which would have been critical for its initial selection and posterior CUG reassignment, are poorly understood. In any case, the appearance of the mutant tRNACAGSer created a unique cellular situation where CUGs were decoded by two distinct tRNAs, namely the new mutant ser-tRNACAGSer and the standard fungal leu-tRNACAGLeu. In the CTG clade ancestor, these two tRNAs competed for approximately 100 My for CUG codons and introduced significant ambiguity at CUG positions [15,16]. The mutant tRNACAGSer was lost in the lineage that originated Saccharomyces spp., maintaining CUG identity for Leu, and was selected in the lineage that originated the CTG clade, thus reassigning CUG identity from Leu to Ser [15].
3 Decoding of CUN codons in Candida spp.
The reassignment of the CUG codon from Leu to Ser affected the decoding properties of Leu CUN codons [17]. In CTG clade species, CUGs are decoded by the tRNACAGSer, while CUA, CUU and CUC codons are decoded by a single tRNA with a 5’-IAG-3’ anticodon (tRNAIAGLeu) (Fig. 2). A single tRNAIAGLeu decodes three different codons as inosine (I) at the first anticodon position base pairs with A, C or U at the third codon position through extended wobble, but the strength of codon-anticodon interactions is variable. The CUU codon is cognate for this tRNA and interacts strongly with it. A similar situation occurs for the 5’-IAG-3’ interaction with the CUC codon however the 5’-IAG-3’-CUA interaction is weak and repressed CUA usage [15]. Conversely, in S. cerevisiae the CUN codon family is decoded by two tRNAs: a tRNAUAGLeu decodes CUA and CUG codons and a tRNAGAGLeu decodes CUC and CUU codons. In S. pombe and in many other fungi the CUN codon family is decoded by three different tRNAs: a tRNAGAGLeu decodes CUC and CUU codons, a tRNAUAGLeu decodes CUA and a tRNACAGLeu decodes CUG codons. Therefore, the appearance of the novel tRNACAGSer influenced CUN decoding and the evolution of Leu tRNAs in the CTG clade species [3,15].
Natural mistranslation of CUGs (∼3% Leu and ∼97% Ser) in extant fungal species of the CTG clade suggests that it may play a role in the biology of these fungi because it elevates the translational error rate by ∼3000-fold (basal error is 10−5) [6,18]. Such a high error rate is highly detrimental in non-adapted species and should have been eliminated by natural selection. Recent studies provide further indirect evidence for a role of CUG ambiguity as engineered C. albicans strains tolerate up to 28% of Leu misincorporation at CUG codons (28,000-fold increase over typical translational error). This does not have visible effects on growth rate, but rather impacts on genome stability and phenotypic diversity [4,19]. Indeed, hypermistranslator strains of C. albicans display a diverse array of forms, up regulate lipase and proteinase secretion and floculate [19]. These cells also generate population heterogeneity, produce aerial hyphae and have high frequency of white-opaque switching and often form long filaments [19]. Interestingly, morphological variation, yeast-hypha transition, proteinase and lipase secretion and adhesins, are important Candida spp. virulence traits [20], suggesting that CUG ambiguity may be relevant for adaptation.
4 The proteomic relevance of CUG reassignment and ambiguity
The global impact of CUG ambiguity is better comprehended if one analyses the genome distribution of CUGs. The C. albicans genome encodes 26,148 CUGs distributed over 66% of its genes at a frequency of 1 to 38 CUGs per gene. The majority of the genes contain between 1 to 5 CUGs (57.7%) (Fig. 3A). Considering the insertion of Ser and Leu at each of these CUG positions the number of protein molecules that can be produced from C. albicans genes is given by the expression 2n, where n = number of CUGs per gene (Fig. 4 for the case n = 3). A genome--wide analysis of CUGs showed that C. albicans has the capacity to synthesize 283 billion different protein molecules from its 6438 genes (Fig. 3B) [4]. In other words, mistranslation of each C. albicans gene produces an array of protein molecules containing Leu or Ser at CUG positions, creating heterogeneous populations of protein molecules differing in Leu or Ser at CUG positions [4]. The biological implications of this phenomenon are profound as each C. albicans cell contains a unique combination of protein molecules (the proteome is statistical) and, therefore, the probability of finding two identical cells in a population is extremely small, even if cells are grown under the same conditions and express the same genes. If one takes into consideration the number of molecules per cell for each protein, a picture of extreme proteome complexity emerges. In yeast low and high abundance proteins are represented by 50 and 106 molecules per cell, respectively [21]. Assuming that: (1) all C. albicans genes are expressed; (2) 10% of the proteins with lowest CAI values are represented by 5000 molecules/cell; (3) 10% of the proteins with the highest CAI value are represented by 50,000 molecules/cell; and (4) the remaining 80% of genes are represented by 20,000 molecules/cell [11], then the approximate number of different protein molecules that result from 3% of CUG ambiguity is 6.7 × 106. This value increases up to 10.7 × 106 in cells mistranslating at 5% and in the case of engineered C. albicans strains that mistranslate CUGs at 28% the total number of different protein molecules that can be produced is 42.8 × 106. In other words, the C. albicans proteome is plastic, highly complex, and its size is unrelated to its gene pool by several orders of magnitude. Whether novel functions are associated to mistranslated proteins or whether they simply represent a nuisance to the cell remains to be elucidated.
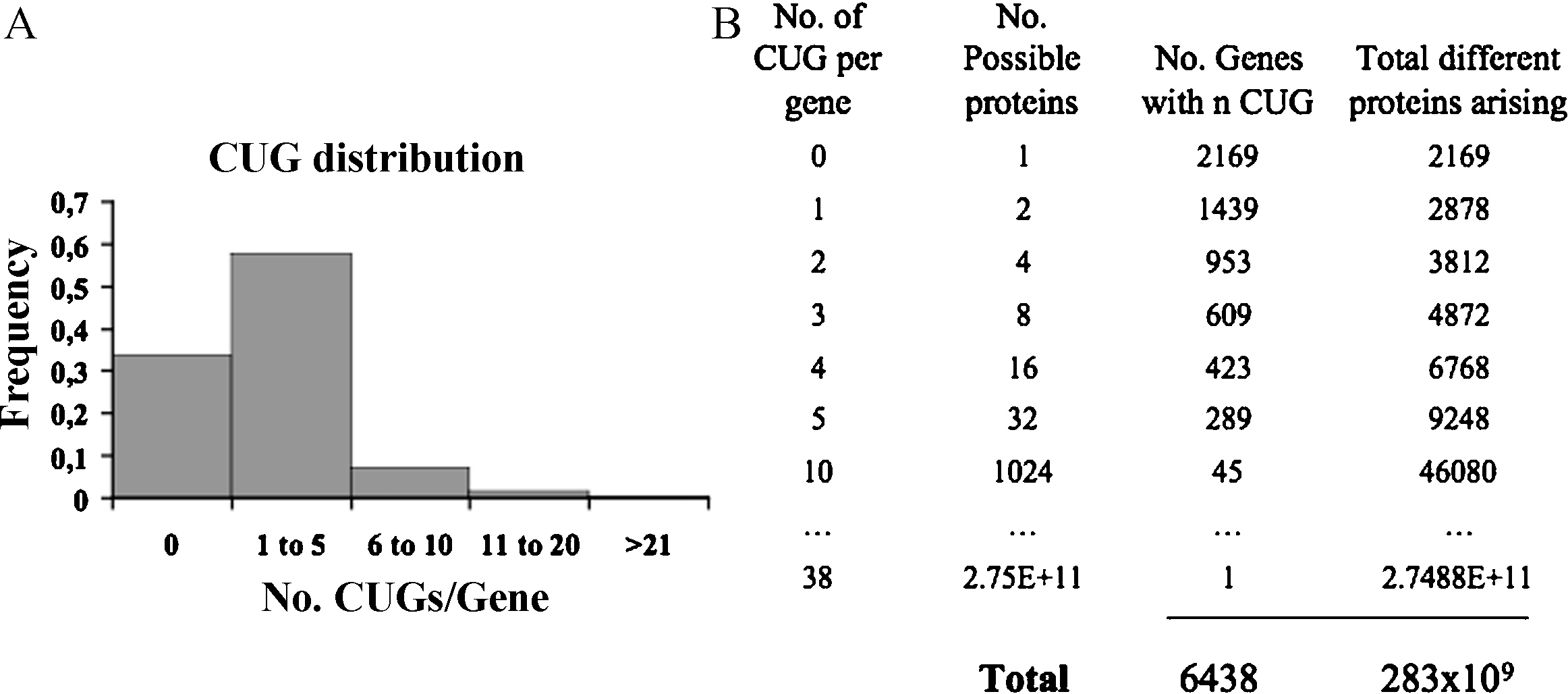
Distribution of CUG codons in C. albicans genes. A. One third of the C. albicans genes do not contain CUG codons. The majority (57.7%) contains between 1 to 5 CUGs and 7.1% have between 6 and 10. Only a small fraction of genes have more than 10 CUG codons. B. Ambiguous CUG decoding expands the C. albicans proteome exponentially. The total theoretical number of combinatorial proteins encoded by the C. albicans genome is 283 billion, considering insertion of Ser (100%) or Leu (100%) at CUGs [6].

Production of combinatorial proteins in C. albicans. The table shows the type of protein molecules that can be produced from a gene containing 3 CUG codons. The probability of synthesis of the protein isoforms depends on the level of leucine incorporation; 3% at 30 °C, 5% at pH 4.0 and 28% in recombinant cells. Leu and Ser insertion at each CUG occurs randomly and is only dependent on the level of ambiguity.
5 CUGs are enriched in specific gene classes
C. albicans CUG usage is repressed in highly expressed genes (high CAI value) and is more relaxed in genes whose expression is low (low CAI value). Indeed, 83% of the highly expressed genes do not have CUGs while 81% of the genes expressed at low level have at least 1 CUG, indicating that CUGs are rarely used (0.43% usage). If one takes into consideration the Specific Codon Usage (SCU), which in this case measures the relative frequency of CUGs normalized to Ser abundance [22], then a clearer picture of CUG usage emerges. The SCUCUG of genes containing one Ser-CUG residue and no other Ser is 1.0 while that of genes containing two Ser-CUGs and 18 additional Ser residues encoded by other Ser codons is 0.1. This allows one to obtain a global picture of CUG usage in C. albicans and identify functional classes of genes enriched in CUGs by carrying out a global survey of SCUCUG values in Gene Ontology (GO) lists (Tables 1 and 2).
Functional categories (GO terms) of genes with highest SCUCUG.
| Gene functional categories | SCUCUG |
| Extrinsic membrane protein (ISS) | 0.094 |
| Golgi to endosome transport (ISS) | 0.090 |
| Spliceosome complex (ISS) | 0.078 |
| DNA repair (IEA) | 0.070 |
| Centromere (ISS) | 0.069 |
| Regulation of redox homeostasis (ISS) | 0.068 |
| Nuclear membrane (ISS) | 0.067 |
| AP-N adaptor complex (ISS) | 0.064 |
| Golgi to vacuole transport (ISS) | 0.063 |
| DNA replication factor complex | 0.062 |
| Chromatin silencing (ISS) | 0.062 |
| SAGA complex (ISS) | 0.061 |
| CCR4-NOT complex (ISS) | 0.060 |
| mRNA splicing (IEA) | 0.059 |
| Spindle pole (ISS) | 0.057 |
| Chromatin (ISS) | 0.057 |
| Golgi to plasma membrane transport (ISS) | 0.057 |
| Mitosis (IEA) | 0.056 |
| Cytokinesis (IEA) | 0.056 |
| Protein targeting (ISS) | 0.055 |
Functional categories (GO terms) of genes with lowest SCUCUG.
| Gene functional categories | SCUCUG |
| Ribosome (ISS) | 0.0057 |
| Cytosolic ribosome (sensu Eukarya) (ISS) | 0.0061 |
| ATP synthesis coupled proton transport (ISS) | 0.0062 |
| Respiratory chain complex | 0.0109 |
| Carbohydrate metabolism (ISS) | 0.0154 |
| Chromatin assembly/disassembly (ISS) | 0.0161 |
| Hydrogen-transporting ATPase | 0.0186 |
| Extracellular (ISS) | 0.0189 |
| Protein biosynthesis (ISS) | 0.0190 |
| Ergosterol biosynthesis (ISS) | 0.0193 |
| DNA-directed RNA polymerase | 0.0203 |
| Proteasome | 0.0209 |
| RNase complex (ISS) | 0.0242 |
| Glycogen metabolism (ISS) | 0.0249 |
| Ubiquitin-dependent protein catabolism (ISS) | 0.0250 |
| Peroxisome | 0.0253 |
| Endocytosis (IEA) | 0.0254 |
| Eukaryotic translation initiation factor complex (ISS) | 0.0261 |
| Heme biosynthesis (ISS) | 0.0264 |
| 35S primary transcript processing (ISS) | 0.0268 |
Plasma membrane protein genes show 2-fold increase in CUG usage (Table 1) and similar positive CUG usage biases were found in genes encoding nuclear membrane proteins and in genes encoding proteins of the SAGA-complex which is a large multi-protein complex with histone acetyltransferase activity involved in transcriptional regulation (ex: Gcn5p; [23]). Conversely, spindle pole protein genes involved in the organization of the cytoskeleton and CCR4-NOT complex genes involved in transcriptional regulation, mRNA degradation and post-transcriptional modifications [24], show negative CUG usage bias, indicating that CUGs are under negative selective pressure in these genes (Table 2). CUG codons are also repressed in ribosomal protein genes, however these proteins are highly expressed and CUG repression is likely related to expression level rather than protein function.
6 Conclusions
Fungal species of the CTG clade use a mutant Ser tRNACAGSer to decode thousands of CUGs as Ser. This tRNA appeared prior to the split between Saccharomyces and Candida genera and competed for over 100 My with the wild type tRNACAGLeu for CUGs during mRNA decoding [15,16]. The Saccharomyces spp. ancestor lost the mutant Ser tRNACAGSer and maintained the standard identity of the CUG codon (Leu), while the ancestor of the CTG clade lost the cognate tRNACAGLeu, retained the mutant tRNACAGSer, and changed the identity of the CUG codon from Leu to Ser. This reassignment resulted in massive mutational change of CUGs to the UUG or UUA Leu codons [15]. Therefore, the CUGs present in extant species of the CTG clade are “new” CUGs that were captured by the tRNACAGSer upon mutation of codons from amino acid families belonging to Ser or amino acids with similar chemical properties [15]. Finally, the double identity of the CUG codon creates statistical proteins and proteomes whose biological implications are still poorly understood. Whether Candida spp. regulate and take advantage of CUG ambiguity remain important questions for future studies.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.