

1 Introduction
A subset of genetic diseases is caused by point mutations that introduce premature stop codons into the coding sequence of genes. These nonsense mutations account for up to 70% of Hurler syndrome cases and 10 to 20% of both muscular dystrophy and cystic fibrosis cases. Induction of stop codon readthrough offers possibilities to reduce the pathology of this type of genetic diseases. Three kinds of factors can promote translational readthrough: drugs acting on the ribosome decoding site such as aminoglycosides, suppressor tRNAs, and factors which decrease translation termination efficiency. However, profound differences exist among these factors in terms of both specificity and efficiency towards suppression of stop codons.
Aminoglycosides have no specificity for stop codons but introduce extensive errors in proteins. Aminoglycosides containing the 2-deoxystreptamine ring bind the small ribosomal subunit RNA, flipping out conserved rRNA residues that are normally displaced upon binding of the cognate tRNA, thus increasing the initial binding affinity of tRNA and decreasing translation accuracy [1]. Even though most of the studies on aminoglycosides were performed in bacteria, it is known for decades that aminoglycosides are also active in eukaryotic cells. Since this first report that aminoglycosides can suppress stop codons in human tissue culture cells [2], numerous studies have assessed in vitro and in vivo the potential of aminoglycosides as a treatment for genetic diseases caused by premature stop codon mutations [3–6]. It has been shown for various genetic diseases such as cystic fibrosis [7] and Duchenne muscular dystrophy [8] that aminoglycosides induced translational readthrough of stop codons and partially restored variable amounts of active full-length proteins. However, the readthrough efficiency is depending on the mutation tested [9], on the nucleotide context of the stop codon [10] and on other unknown factors involved in readthrough regulation [11]. Besides, the efficiency of aminoglycoside therapy depends on the molecular mechanism by which the nonsense mutation causes the defects and on the level of corrected protein required to reduce clinical symptoms [12]. Moreover, although aminoglycosides are widely used as antimicrobial agents, the major limitation to their clinical use is the development of nephrotoxicity and ototoxicity [13]. These toxic side effects could impede long term treatment at the high doses required to induce efficient readthrough in mammalian cells.
Suppressor tRNAs have the advantage of being highly specific for one of the stop codons. The relative frequency of termination versus suppression is determined by parameters such as codon context [14], the activity of termination factors [15], and obviously, the abundance of natural suppressor tRNAs. However, with the exception of selenocysteine tRNA, natural suppressor tRNAs are scarce in mammalian cells and all nonsense suppressors characterized to date are normal cellular tRNAs that are needed for reading their cognate sense codons and are able to recognize stop codons through unconventional codon–anticodon base pairings [16]. However, recognition of stop codons by these natural suppressor tRNAs is quite inefficient. In 1982, it has been shown that an artificial human nonsense suppressor tRNA was able to suppress in vitro the UAG mutation in the β-globin gene mRNA obtained from a patient suffering from β-thalassemia [17]. Efficient nonsense suppression was obtained by transient overexpression of suppressor tRNAs in human cell tissue culture [18]. However, it has been reported that constitutive expression of suppressor tRNAs is detrimental to mammalian cells [19].
In eukaryotes, two release factors, eRF1 and eRF3, are required to terminate protein synthesis. eRF1 recognizes all three stop codons within the ribosomal A site and triggers the hydrolysis of the peptidyl-tRNA, generating a free full-sized polypeptide [20]. eRF3 is a GTPase that stimulates eRF1 activity and confers GTP dependency upon the termination process [21]. Recently, it has been shown that small interfering RNAs (siRNAs) targeting translation termination factor mRNAs increase translational readthrough in mammalian cells [22–24]. Thus, it has been proposed that synthetic siRNAs could be used as therapeutic agents in diseases caused by nonsense mutations [24]. Indeed, numerous studies have reported the potential for using siRNAs as therapeutic agents and several preclinical works relating to the treatment of cancers and infections have now been published [25].
In this study, we compared the ability of various aminoglycosides, other types of antibiotics acting on the ribosome, suppressor tRNAs and siRNAs targeting translation termination factor mRNAs to promote stop codon readthrough in human cells. We used a modified 293 cell line stably expressing a lacZ gene containing a premature stop codon. The synthesis of full-length active β-galactosidase required stop codon readthrough, and thus, the level of β-galactosidase activity reflected readthrough efficiency. Our results show that the highest level of readthrough was obtained with suppressor tRNAs. However, they also show that high levels of readthrough were obtained upon prolonged gentamicin treatment, whereas prolonged expression of siRNA targeting eRF3a mRNA only moderately increased readthrough. In view of these results, we discuss the potentials for these three kinds of factors to suppress disease-associated premature stop codon mutations.
2 Materials and methods
2.1 Antibiotics
Gentamicin sulfate, hygromycin B and paromomycin sulfate, erythromycin and chloramphenicol were obtained from Sigma. Netilmicin (Nétromicine) was purchased from Schering-Plough, amikacin (Amiklin) from Bristol-Myers-Squibb and tobramycin sulfate (Nebcine) from Erempharma.
2.2 Plasmids
Plasmids expressing small interfering RNAs si-3a1 (targeting eRF3a mRNA) and si-1Y (targeting eRF1 mRNA) were constructed using pSuper vector as previously described [23]. Plasmids ptRNAam and ptRNAoc expressing amber and ochre suppressor tRNA, respectively, have been previously described [15].
2.3 Cell culture, electroporation and drug treatment
The 559C cell line is a derivative of human 293 cell line (ATCC N○ CRL-1573) stably expressing a lacZ gene in which the coding sequence is interrupted by a TAG stop codon [23]. 559C cells were maintained in Dubelcco modified Eagle's minimum essential medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum, 100 μg/ml streptomycin and 100 units/ml penicillin. For drug treatment, 100-mm-diameter plates of 559C cells at 50% confluency, were washed with 10 ml of PBS and incubated at 37 °C with 10 ml of DMEM supplemented with 10% fetal calf serum (lacking streptomycin and penicillin) and drugs at concentration indicated in the text and figure legends. After treatment, the medium was removed and cells were collected by scraping in 10 ml of PBS, and pelleted. The electroporation was performed as described [23] with a Gene pulser II electroporation system (Bio-Rad) using cells and 10 μg of plasmid DNA. The medium was changed the day after electroporation, and cells were collected two to five days later by scraping in 10 ml of PBS, and pelleted.
2.4 Readthrough assay
Readthrough assay was described in [23]. Briefly, cell pellets resuspended in 300 μl of 100 mM Sodium phosphate buffer, pH 8 were lysed by four cycles of freezing and thawing and cell lysates were centrifuged at 12 000 g for 30 min at 4 °C. Supernatants were assayed for total proteins using the Micro BCA Protein Assay Reagent Kit (Pierce) and β-galactosidase activity using the Luminescent beta-galactosidase Detection Kit II (Clontech). For each sample, total protein concentration was determined using the Micro BCA Protein Assay Reagent Kit (Pierce), bovine serum albumine being used as standard. The β-galactosidase assays were performed using the Luminescent beta-galactosidase Detection Kit II (Clontech) and following the manufacturer instructions. The β-galactosidase activity was measured as relative light units (R.L.U.) using a single photon count program on scintillation counter. The β-galactosidase activity was expressed as R.L.U./μg of total proteins and the readthrough efficiencies were calculated as sample activity relative to negative control activity (negative controls are mentioned in the figure legends).
3 Results
3.1 Comparison of the readthrough efficiency of various aminoglycosides
Gentamicin was commonly used for studies on aminoglycosides-mediated rescue of disease-causing nonsense mutations. However, a few studies have compared the efficiency of other aminoglycosides to induce stop codon readthrough [26–28]. In a first set of experiments, we tested five aminoglycosides of the 2-deoxystreptamine family that are routinely used for clinical purposes (gentamicin, amikacin, netilmicin, tobramycin and paromomycin) and hygromycin B which has a 5-substituted 2-deoxystreptamine ring and is not used in human therapy. In contrast to the typical 2-deoxystreptamines which cause misreading by interfering with normal binding of A site-tRNA, hygromycin B interferes with the translocation step by affecting the EF-2-mediated transfer of A-site-tRNA to the P site [29]. The efficiency of stop codon readthrough was measured in human cells stably expressing a lacZ reporter gene interrupted by a premature nonsense TAG codon and encoding a truncated inactive β-galactosidase – 559C cell line. The synthesis of full-length active β-galactosidase required stop codon readthrough and thus, the level of β-galactosidase activity reflected readthrough efficiency. For each aminoglycoside, the working concentration was determined empirically in order to provide maximum readthrough efficiency without induction of cell death (loss of cell adherence). Parallel cultures of 559C cells were treated for 48 hours with individual aminoglycosides at working concentration and the β-galactosidase activity (relative light units/μg of protein) was measured in cell extracts. The relative readthrough efficiency was calculated by dividing the β-galactosidase activity in each sample by the β-galactosidase activity in the extract of non-treated cells (Fig. 1). A clear increase in readthrough was observed for cells treated with gentamicin (1 mg/ml), hygromycin B (0.1 mg/ml) and amikacin (3 mg/ml), whereas netilmicin (1 mg/ml), tobramycin (1 mg/ml) and paromomycin (2 mg/ml) had no effect. Gentamicin increased readthrough by ∼4-fold, a level in good agreement with the results reported by others [10]. A comparable increase in readthrough of ∼3.4-fold was observed with amikacin, whereas the highest readthrough level (∼10-fold) was obtained with hygromycin B treated cells.
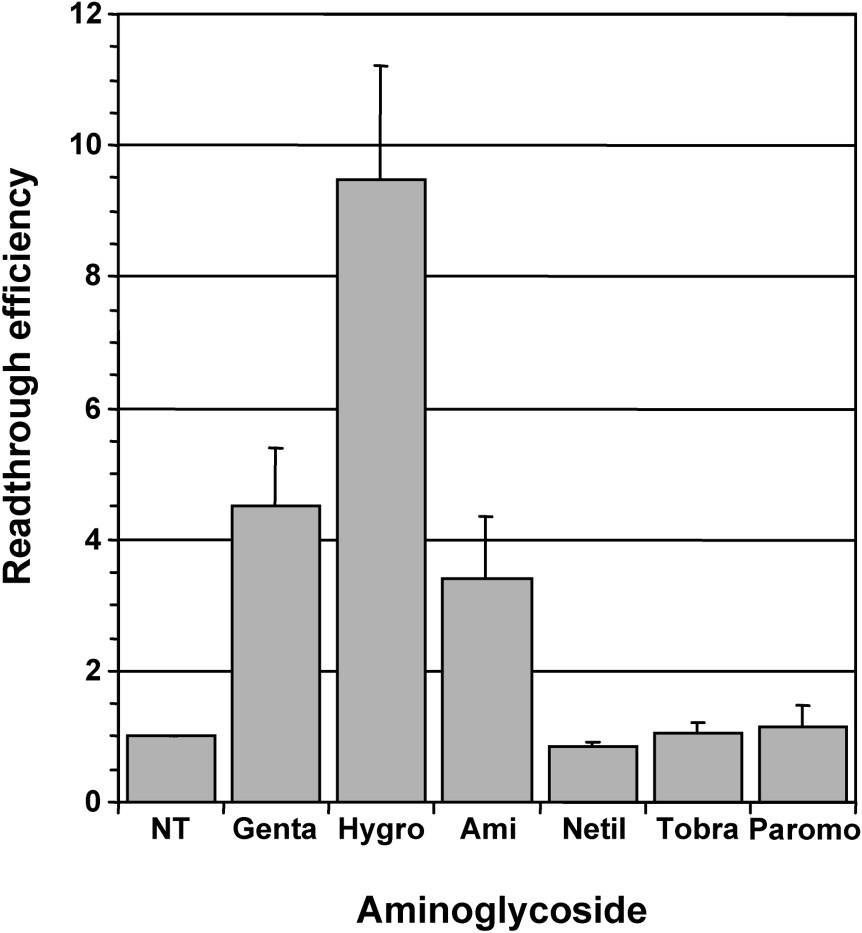
Comparison of readthrough induced by various aminoglycosides. Parallel cultures of 559C cells stably expressing a lacZ gene containing a premature UAG stop codon were treated for 48 hours with 1 mg/ml gentamicin (Genta), 0.1 mg/ml hygromycin B (Hygro), 3 mg/ml amikacin (Ami), 1 mg/ml netilmicin (Netil), 1 mg/ml tobramycin (Tobra) and 2 mg/ml paromomycin (Paromo); non-treated cells (NT) served as negative control. The β-galactosidase activity (relative light units/μg of protein) was measured in cell extracts. The relative readthrough efficiency was calculated by dividing the β-galactosidase activity in each sample by the β-galactosidase activity in the extract of non-treated cells. Results were expressed as the mean of four experiments, error bars show the standard error of the mean.
We then compared the effect of gentamicin, which was used as reference for aminoglycosides, with other antibiotics targeting the large ribosomal subunit, i.e., chloramphenicol which is known to block peptidyl transferase activity by hampering the binding of tRNA to the ribosome A site, and macrolides of the erythromycin class which do not affect peptidyl transferase activity, but block the entrance to the tunnel that guides the nascent peptides away from the peptidyl transferase centre [30]. As shown on Fig. 2, neither chloramphenicol nor erythromycin affect the readthrough level when added to the culture medium of 559C cell. Moreover, when tested in association with gentamicin, they did not enhance the effect observed with gentamicin alone (Fig. 2). These results suggest that chloramphenicol and macrolides: (i) did not induce translational errors in the eukaryotic ribosome; and (ii) did not potentiate gentamicin effect on the ribosomal decoding site.
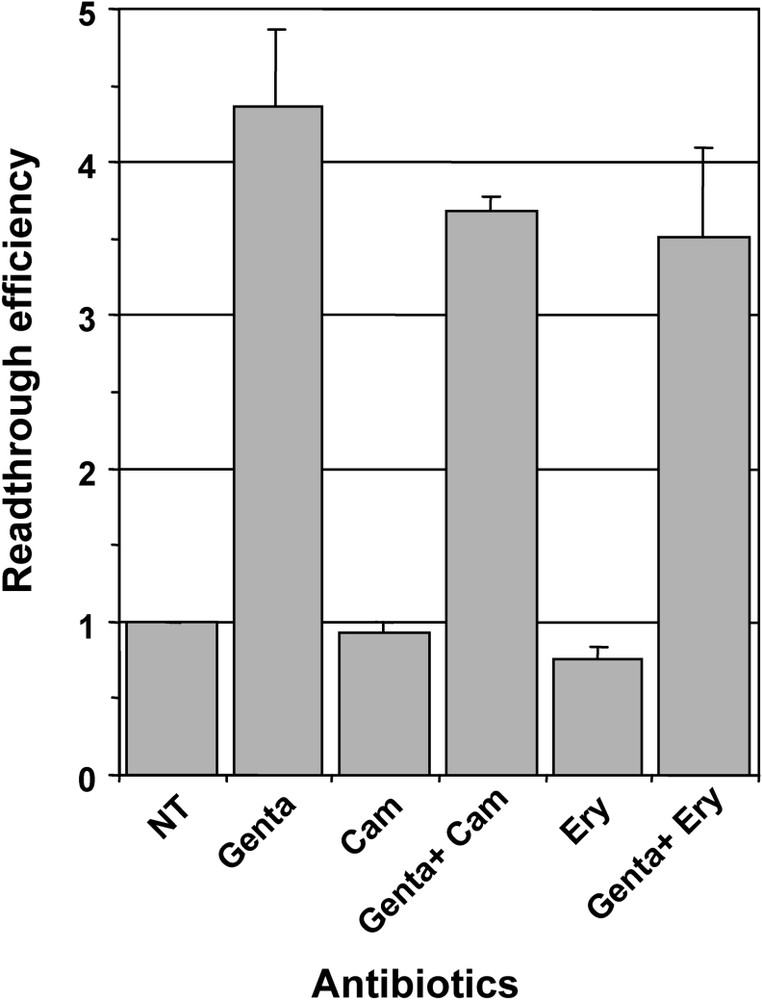
Effect of chloramphenicol and erythromycin on readthrough induced by gentamicin. Parallel cultures of 559C cells were treated for 48 hours with 1 mg/ml gentamicin (Genta), 20 μg/ml chloramphenicol (Cam), 1 mg/ml gentamicin plus 20 μg/ml chloramphenicol (Genta + Cam), 0.5 mg/ml Erythromycin (Ery) and 1 mg/ml gentamicin plus 0.5 mg/ml Erythromycin (Genta + Ery); non-treated cells (NT) served as negative control. The β-galactosidase activity was measured in cell extracts and the relative readthrough efficiency was calculated by dividing the β-galactosidase activity in each sample by the β-galactosidase activity in the extract of non-treated cells. Results were expressed as the mean of two experiments, error bars show the standard error of the mean.
3.2 Comparison of the readthrough efficiencies of aminoglycosides, siRNA targeting release factor mRNAs and suppressor tRNAs
In addition to their high toxicity for both kidney and inner ear, aminoglycosides cause codon misreading at sense and stop codons hence increasing the ribosome error rate leading to the synthesis of numerous abnormal proteins. Thus, we compared the readthrough efficiencies of aminoglycosides with those of suppressor tRNAs and siRNAs targeting release factor mRNAs which offer the advantage to specifically induce misreading at stop codons. For this purpose, plasmids expressing siRNAs specifically targeting either eRF1 (si-1Y) or eRF3a (si-3a1) and plasmids encoding ochre (UAA) and amber (UAG) suppressor tRNA genes were electroporated in 559C cells. As we previously showed that the readthrough efficiency of siRNAs targeting release factors is maximum at 5 days post-electroporation [23], cells were treated for 5 days with 1 mg/ml of gentamicin in order to obtain comparable results (notice that the medium with gentamicin was changed every two days). As shown in Fig. 3, the cognate amber (UAG) suppressor tRNA very efficiently suppressed the UAG stop codon in the lacZ reporter mRNA, whereas the non-cognate ochre (UAA) suppressor tRNA had no effect. The levels of readthrough obtained with the siRNAs targeting eRF1 and eRF3a (∼8-fold and ∼40-fold the basal level, respectively) are roughly the same as those previously published [23]. Interestingly, the increase in readthrough was about 20-fold higher in cells treated for 5 days with gentamicin than that measured in cells after two days of treatment: ∼85-fold (Fig. 3) versus ∼4-fold (Fig. 1). This result suggests that a relatively high level of readthrough can be obtained upon prolonged gentamicin treatment. Indeed, we did observe a large increase in readthrough (up to 300-fold the level of non treated cells) when cells were treated for a week with gentamicin. However, prolonged gentamicin treatments induced an arrest of cell growth. As our reporter lacZ system is not sensitive to the nonsense mediated decay pathway of RNA degradation as shown by the absence of modification of the steady state lacZ mRNA level when depletion of eRF3a increased the readthrough level [23], the large increased in readthrough can not account for an increased in mRNA stability. However, the accumulation of β-galactosidase and its stability could be some of the determinants for this effect.
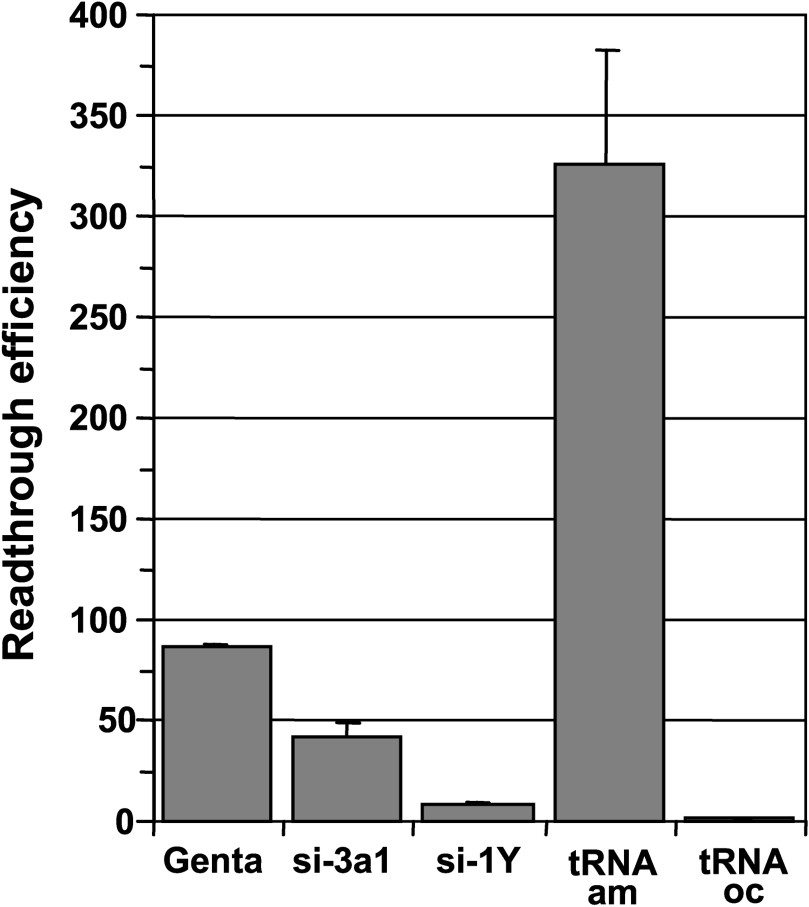
Comparison of gentamicin, release factor targeted siRNAs and suppressor tRNAs readthrough efficiencies. 559C cells stably expressing a lacZ gene containing a premature UAG stop codon were either treated with 1 mg/ml gentamicin (Genta) or electroporated with the empty vector pSuper, with plasmid expressing siRNAs directed against eRF3a (si-3a1), or eRF1 (si-1Y), and with plasmid expressing amber suppressor tRNA (tRNAam) or ochre suppressor tRNA (tRNAoc). Cell extracts were performed 5 days after electroporation and used for β-galactosidase assay. Readthrough efficiencies were calculated by dividing the β-galactosidase activity in each sample by the β-galactosidase activity in pSuper-electroporated cell extract. Results were expressed as the mean of three experiments, error bars show the standard error of the mean.
3.3 Effect of prolonged depletion of release factors
We next determined whether a prolonged depletion of release factors could induce high readthrough levels. To this end, 559C cells were depleted in either eRF3a or eRF1 by repeated electroporation of the plasmid expressing either si-3a1 or si-1Y, respectively. Cells electroporated with plasmid pSuper served as a negative control. Five days after the first electroporation, cells were collected, and cells were re-electroporated with the same plasmid, i.e., either pSuper vector, plasmid expressing si-3a1 or plasmid expressing si-1Y. The remaining cells were used for β-galactosidase assays. Using the same experimental procedure, five days after re-electroporation, cells were electroporated again a third and a fourth time with the same plasmids. The effect of the multiple electroporations on stop codon readthrough was analysed by measuring the β-galactosidase activity in cell extracts. For each electroporation, readthrough efficiencies of siRNAs were calculated by dividing the β-galactosidase activity in each sample by the β-galactosidase activity in pSuper-electroporated cell extract. As shown in Fig. 4, the readthrough level induced by si-3a1 targeting eRF3a increased moderatly after the second electroporation, reached a maximum after the third electroporation (∼2-fold the level of the first electroporation), and then decreased after the fourth electroporation. Comparable increase and decrease in readthrough levels were observed with si-1Y targeting eRF1. However, with a maximum of ∼12-fold, the relative readthrough levels induced by si-1Y were much lower than those obtained with si-3a1. The decrease in readthrough observed after the fourth electroporation could be due to the deleterious effect of either readthrough or prolonged release factor depletion on cell growth. Both of these possibilities are supported by the fact that cells repeatedly electroporated with plasmids expressing si-3a1 and si-1Y grew slower than those electroporated with the empty pSuper vector. Since the level of readthrough is low in si-1Y electroporated cells, a global effect of readthrough on protein synthesis is unlikely to be responsible for the observed decrease. Another possibility is that eRF1 and eRF3a depletion generates other effects than translational readthrough which could affect cell growth. Nevertheless, these results showed that the readthrough level increased moderately (∼100-fold) when cells were depleted in eRF3a for 15 days (three repeated electroporation). As a comparison, higher levels of readthrough (∼300-fold) than those observed with prolonged eRF3a depletion were obtained either after 7 days of gentamicin treatment or 5 days after electroporation of the plasmid expressing the amber suppressor tRNA.
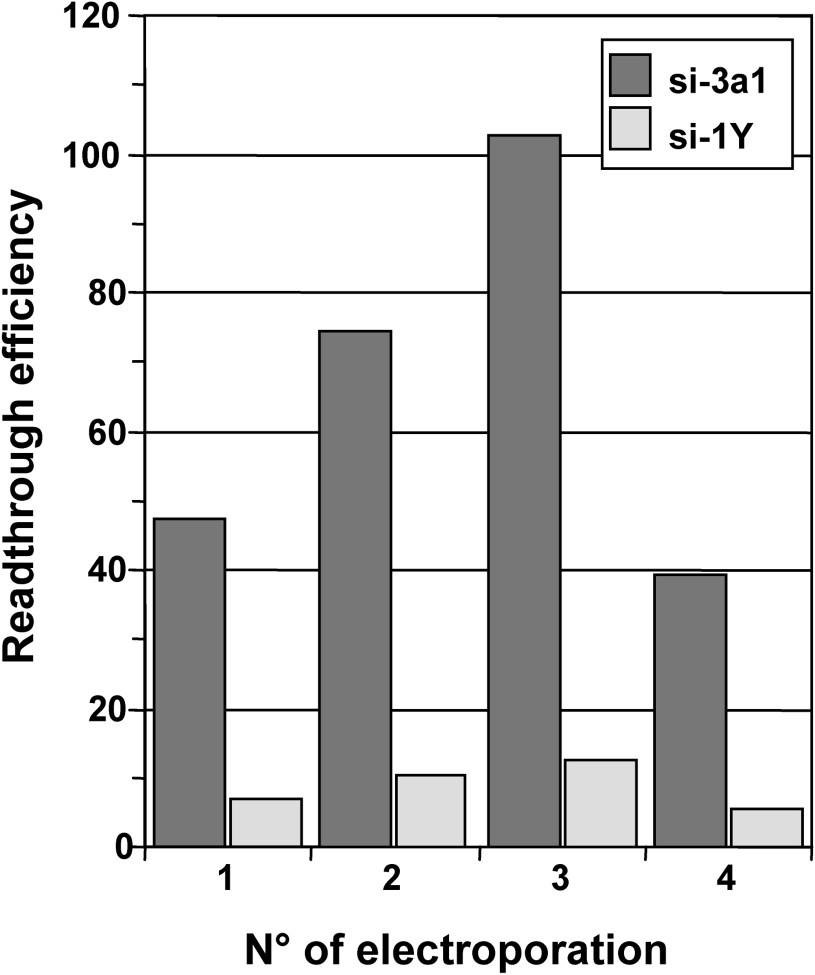
Effect of prolonged eRF1 and eRF3a depletion on stop codon readthrough. Parallel 559C cell cultures were electroporated either with the empty vector pSuper or with pSuper derivatives expressing siRNAs directed against eRF3a (si-3a1) or eRF1 (si-1Y). Five days after the first electroporation (electroporation No. 1), cells were collected, a fraction was re-electroporated with the same plasmid (electroporation No. 2) and the remaining fraction was used for β-galactosidase assay. This procedure was repeated two more times (electroporation No. 3 and 4). Readthrough efficiencies of siRNAs were calculated by dividing the β-galactosidase activity in each sample by the β-galactosidase activity in pSuper-electroporated cell extract.
4 Discussion
We report here a comparative study of translational readthrough efficiencies induced by aminoglycosides, suppressor tRNAs and translation termination factor depletion in human tissue cell culture. For this purpose, we measured the readthrough at a nonsense UAG codon interrupting a lacZ reporter gene stably expressed in the human 293 cell line. Although it has been shown in a systematic analysis that the aminoglycoside-induced readthrough is greater at UGA codon than at UAG or UAA [10], the statistics for nonsense mutations in the Human Gene Mutation Database reveal that the UAG codon is the most frequent nonsense mutation causing genetic disease encountered: UAG account for 40.4%, UGA 38.8% and UAA 20.8% of the ∼7000 nonsense mutations collected to date [31].
Among the classical 2-deoxystreptamine aminoglycosides tested in this study, only gentamicin and amikacin clearly increased the readthrough level (Fig. 1). This result is in good agreement with the result of Keeling and Bedwell [26] showing that, with an identical pattern of context dependence, amikacin stimulated readthrough in a way similar to gentamicin. In addition, it has been reported that amikacin has lower nephrotoxic side-effects than all 2-deoxystreptamine-containing aminoglycosides tested so far [32]. Therefore, we agree with the conclusion of Keeling and Bedwell [26] that amikacin represents an alternative to gentamicin for suppression therapy.
In our readthrough assay, the highest level of readthrough was obtained with hygromycin B (∼2-fold the level obtained with gentamicin, Fig. 1). This is surprising because it has been reported that, in eukaryotes, hygromycin B affects ribosomal translocation and has low misreading potency [29]. However, those results were obtained in vitro using Tetrahymena ribosomes. Our results suggest that, in human cultured cells, the block of ribosomal translocation caused by hygromycin B induced an increase in readthrough. Hygromycin B is not used in clinical medicine mainly because of its activity on eukaryotic translation. Our data suggest that the potential of hygromycin B as a therapeutic agent for genetic diseases caused by premature nonsense mutations should be reconsidered.
We also observed that high levels of readthrough can be obtained when cells are treated for 5 to 7 days with gentamicin. This suggests that in addition to the dosage, the duration will be an important parameter for aminoglycosides treatment. A course of treatment which should employ the lowest effective dose for the minimum length of time necessary for effective treatment, will depend on the efficiency of readthrough required for obtaining the amount of active full-length protein that will relieve, at least partially, the disease symptoms. The major drawback of aminoglycosides is their nephrotoxicity and ototoxicity. However, it was observed that ototoxicity and nephrotoxicity did not correlate (see for review [13]). The nephrotoxic effect of aminoglycosides is reversible and is correlated to the accumulation of these drugs in the cortex of the kidney. Thus, the duration of exposure to aminoglycosides is a critical factor since it determines the extent of renal drug uptake. In contrast, ototoxicity is usually irreversible. Moreover, it was demonstrated that cochlear damage induced by gentamicin was not associated with an accumulation of the drug into the inner ear. Because aminoglycoside accumulation in the ear is dose-dependent but saturable, the dosage regimen of aminoglycosides is an important determinant of the ototoxicity. Thus, the choice of the aminoglycoside, its dosage regimen and the duration of exposure will determine the level of readthrough that can be obtained and will greatly influence treatment efficacy.
The comparison of aminoglycoside treatment with suppressor tRNA expression and translation termination factor depletion reveals the high efficiency of suppressor tRNAs. Unfortunately, it has been reported that constitutive expression of suppressor tRNAs are toxic to mammalian cells [19]. However, inducible systems have been constructed in which, the activity of the suppressor tRNAs was dependent upon the concomitant expression of a tetracycline-regulated E. coli glutaminyl-tRNA synthetase [33]. In these systems, regulated suppression of stop codons could prevent the deleterious effect of suppressor tRNA constitutive expression in mammalian cells. Nevertheless, the use of suppressor tRNA systems requires: (i) identification of the stop codon which can be difficult in long mRNAs; and (ii) effective delivery and appropriate tissue distribution of the two genes in the case of inducible systems.
Concerning the readthrough induced by translation termination factor depletion using siRNAs, profond differences were observed between eRF1 and eRF3a (Figs. 3 and 4). Only a low level of readthrough was obtained by expression of eRF1 mRNA-targeted siRNA. This low readthrough efficiency is probably due to the partial depletion of eRF1 as previously reported with either various siRNAs or antisense oligonucleotides [24]. Conversely, we have previously shown that the expression of siRNA 3a1 targeting eRF3a mRNA induced a profound depletion of eRF3a five days after electroporation. However, eRF3a depletion induced only a moderate increase of readthrough (∼40 fold) when compared to the level obtained with amber suppressor tRNA (∼350-fold) and gentamicin (∼80-fold). In addition, the level of readthrough increased only 2-fold when cells were submitted to repeated electroporations (Fig. 4). This difference could be explained by the fact that in the case of eRF3a depletion, the readthrough at the UAG stop codon requires decoding of UAG by natural suppressor tRNAs that are near cognate tRNAs. However, the mechanism of amino acid incorporation by near cognate tRNAs is intrinsically inefficient [16]. In the case of cells treated with aminoglycosides, decoding of the UAG stop codon by near and non cognate tRNAs is greatly enhanced by the presence of the aminoglycoside molecule in the ribosomal decoding site, i.e., the flipping out of the two bases of the 18S rRNA which mimics cognate tRNA binding. Conversely, the high efficiency of readthrough in cells electroporated with the amber suppressor tRNA gene, is explained by the fact that the UAG codon is efficiently decoded by a cognate tRNA and by the high amount of suppressor tRNA expressed from the plasmid.
Although eRF3a depletion induced a moderate readthrough level, the use of siRNA has at least two advantages: (i) as for aminoglycoside treatment, the identification of the type of the premature stop mutation is not a prerequisite for the treatment; (ii) conversely to aminoglycosides, eRF3a depletion induces mis-reading at stop codons only. As judged by the number of preclinical work reporting the use of synthetic siRNAs as therapeutic agents, siRNA could become a favoured method for the treatment of genetic diseases caused by premature nonsense mutation. However, the importance of siRNAs off-target effect is far from being accurately assessed.
In fine, despite their side effects and lack of specificity for stop codons, aminoglycosides are to day the simplest way for the therapy of disease caused by premature stop codons. As judiciously observed by Kerem [34] talking about gentamicin treatment of cystic fibrosis patients: “The future goal is to maximize the effect of stop-codon suppressors… while minimizing side effects, but further studies must be performed to find a safer compound that may be administered in small children from the time of diagnosis.”
Acknowledgements
This work was supported by the ‘Association française contre les myopathies’ and by the ‘Association pour la recherche sur le cancer’ (grant n○ 3784). Dialo Diop held a fellowship from the ‘Association française contre les myopathies’. Céline Chauvin held fellowships from the French ‘Ministère de la Recherche et de l'Enseignement supérieur’, from the ‘Fondation pour la recherche médicale’ and from the ‘Société française du cancer’.