

1 Introduction
Whereas the ancestor of Opisthokonts was presumably very intron rich, with an intron density estimated of 3.70 or 5.15 introns per kb of coding sequences depending on the evolutionary model used [1,2], modern species vary in their number of introns by orders of magnitude: from a couple of hundred introns per genome in the model yeast Saccharomyces cerevisiae to more than 8 introns per gene in human [3]. The fungal lineage has undergone a striking evolution illustrating the different evolutionary scenarios observed for most of the eukaryotic phylum [1]. In Basidiomycetes such as Cryptococcus neoformans (5.3 introns per gene; [4]), intron gain predominates whereas in most filamentous Ascomycetes, gain and loss rates are balanced to manage an average density of 2.37 introns per kb ([1], [5]). With only few intron-containing genes, hemiascomycetous yeasts represent a clear example of a dramatic reduction of intron number arising from extensive loss dominating over non-significant gains. Mechanistic models proposed for intron loss include homologous recombination between the genomic copy of the gene and a partial cDNA reverse transcribed from the 3’ end of mRNA [6,7]. This mRNA-mediated model is generally admitted for intron loss in fungi where numerous cases are evidenced by comparative genomics of closely related species [5,8]. Loss of splicing due to splice site degeneration or de-intronization are rarely reported for protein-coding genes in these eukaryotes [9] but has been proposed to explain the loss of snoRNA-containing introns in Hemiascomycetes [10]. However, the evolutionary forces and the genetic context leading to the extensive loss of yeast introns in protein-coding genes or noncoding RNAs are still to be understood.
Correlations have been established between intron density and intron structure or distribution. In intron-poor genomes, introns are biased toward the 5’ ends of genes, and this is particularly pronounced in S. cerevisiae [11,12]. The currently accepted explanation invokes replacement of genomic copies by retrotranscribed mRNA and preferential loss of 3’ introns then results from a 3’ bias of reverse transcriptase that often terminates prematurely [7,13]. Compact genomes with few introns tend also to show strong conservation of intron boundaries especially at the 5’ splice site (5'ss) and branching point (BP) [14]. Indeed in S. cerevisiae and at least in some other hemiascomycetous yeasts, a 6-bp sequence at the 5'ss and a 7-bp sequence at the BP are required for efficient splicing [15–17] which is coherent with an intron definition mechanism. In contrast, the polypyrimidine tract (PPT) usually found in metazoan transcripts close to their 3’ splice site (3'ss) is weak or even lacking in yeasts. All these splicing signals seem to have coevolved with their corresponding splicing factors, especially U2AF1 (U2AF59 in S. pombe), U2AF2 (U2AF25 in S. pombe, MUD2 in S. cerevisiae) and SF1, that bind to the 3’ end of introns, i.e. to 3'ss, PPT and BP, respectively, and thus participate in the early-step of pre-mRNA splicing [18–21].
The tenth anniversary of the first report on comparative genomics of hemiascomycetous yeasts [22] is a great opportunity to assess the findings derived from the systematic sequencing of these intron-poor genomes in terms of intron structure, intron dynamics and evolution. We have examined whether the general trends reported for S. cerevisiae or for small sets of introns in other species [16] are conserved along the hemiascomycetous phylum.
2 Materials and methods
2.1 Collecting genome data
The genome sequences and proteomes of fully sequenced genomes were downloaded from the following websites: Candida genome database (http://www.candidagenome.org/) for Candida albicans assembly 21 (strain SC5314), https://bioinformatics.psb.ugent.be/gdb/pichia/ for Pichia pastoris (strain GS115), SGD (http://www.yeastgenome.org/) for Saccharomyces cerevisiae (strain S288c), AGD (http://agd.vital-it.ch/Ashbya_gossypii/index.html) for Eremothecium gossypii (strain ATCC 10895), GeneDB (http://old.genedb.org/genedb/pombe/) for Schizosaccharomyces pombe, Génolevures (http://www.genolevures.org/download.html) for Candida glabrata (strain CBS 138), Zygosaccharomyces rouxii (strain CBS 732), Lachancea (Saccharomyces) kluyveri (strain CBS 3082), Lachancea (Kluyveromyces) thermotolerans (strain 6340), Kluyveromyces lactis (strain CLIB 210), Debaryomyces hansenii (strain CBS 767) and Yarrowia lipolytica (strain E150). Two genomes newly sequenced and annotated by the Génolevures consortium were included in our analysis: Arxula adeninivorans (strain LS3) and Pichia sorbitophila (strain CBS 7064).
The number of introns in S. cerevisiae was corrected according to Juneau et al. [23] and that of L. kluyveri according to Payen et al. [24]. Revisiting the ribosomal protein families based on the Génolevures families and the Ribosomal Protein Gene database [25], we identified 11 genes not previously annotated and modified 2 gene models for start position or intron coordinates. Here are the coordinates of these genes: CAGL0E02013 g (Cagl0E: complement (join(199494.199942, 200692.200803)), CAGL0I03971 g (Cagl0I: complement (350003.350080)), ZYRO0C06963 g (Zyro0 C: complement (525633.525836)), ZYRO0C09493 g (Zyro0 C: 721028.721105), ZYRO0C12111 g (Zyro0 C: complement (join (949177.949326, 949441.949446))), ZYRO0D15697 g (Zyro0D: complement (1314416.1314595)), ZYRO0F12562 g (Zyro0F: complement (join(1020500.1021017, 1021163))), KLTH0F03883 g (Klth0F: join(347007.347012,347389.347538)), KLTH0H02409 g (Klth0H: complement(216579.216656)), KLLA0B07590 g (Klla0B: complement(join(662543.662692, 663246.663251))), KLLA0F05412 g (Klla0F: complement (533118.533195)), SAKL0F10109 g (Sakl0F: 777073.777150), DEHA2D15389 g (Deha2D: complement (1282952.1283029)).
2.2 Phylogeny
A phylogenetic tree was built from the alignment of 101 proteins using the MAFFT algorithm [26] and further cleaned with Gblocks [27]. Cleaned alignments covering more than 75% of the initial alignment were concatenated (84 protein alignments, 6184 resulting amino acids). The tree was constructed by maximum likelihood using PHYML [28] with a JTT substitution model corrected for heterogeneity among sites by a Γ-law distribution using 4 different categories of evolution rates. The proportion of invariable sites and the α-parameter of the Γ-law distribution were optimized according to the data. Bootstrap values were calculated with 100 replicates.
2.3 Intron pattern construction
We developed a Python algorithm that predicts the putative BP from the different introns found in each species. This algorithm is based on the consensus motifs already reported for S. cerevisiae or for other hemiascomycetous yeasts [16,29] and on degenerate motifs found during the step of manual genome annotation. Data on 5'ss, 3'ss and BP are represented as sequence logos [29,30]. Data on S1 (5'ss to BP motif) and S2 (BP motif to 3'ss) distances as well as intron size are collected and showed as minimal, maximal, mean and average sizes. Complete data are available on the Génosplicing splicing website at http://genome.jouy.inra.fr/genosplicing.
3 Results and discussion
3.1 Correlation between intron loss and molecular evolution
The number of introns and intron-containing genes was collected for fully sequenced genomes. We have to emphasize that collecting such data is not straightforward as in some species, sequencing errors or pseudogenes lead to gene models composed of exons separated by artefactual introns. In filamentous fungi, genome annotation is facilitated by transcriptomic data (cDNA sequencing or RNAseq). Unfortunately, gene model prediction in budding yeasts suffers from the lack of this type of experimental data.
The percentage of intron-containing genes in hemiascomycetous yeasts varies from 2.4% in C. glabrata to about 15% in Y. lipolytica [31] which corroborates the fact that hemiascomycetous genomes are intron-poor. We can observe in Fig. 1A that species of the same clade tend to have a comparable percentage of introns (about 5% in protoploids, 6% in CTG species, and more than 10% in early-branching species), even if branch specific evolution also occurred. For instance, among the protoploid species K. lactis has lost about 30% more introns than the other species. An even clearer correlation can be established with the molecular evolution of the species. The correlation represented by the tendency curve on Fig. 1B was validated by a Spearman test (pvalue = 3.8e-07, Rho = −0.8155246). The more rapidly the genomes evolve, the more rapidly they lose their introns. Will the yeasts lose all their introns? Probably not. Some species, like Lachancea (Kluyveromyces) thermotolerans [32], have lost all active retrotransposons and thus all known sources of reverse transcriptase indispensable for cDNA-mediated intron loss: they are probably bound to conserve their introns. But even if they harbour active retrotransposons, yeast species may find it advantageous to keep some of their introns at least. Parenteau et al. [33] showed that most of S. cerevisiae introns can be suppressed with minor consequences. However, among the 87 tested genes three of them required introns for normal growth and all three encoded RNA-binding proteins involved in mRNA metabolism or RNA transport. Additional reasons may explain why some introns are still present and necessary in yeasts: the presence of snoRNA or cis-regulatory containing elements within the introns or mechanisms for meiosis-dependent splicing [23]. Moreover, Juneau et al. [34] reported that introns improve transcriptional and translational yield, confer fitness to yeasts and increase their competitive growth in natural environment. This is however to balance with the fact that rapidly regulated genes in stress conditions are intron-poor in S. cerevisiae [12].
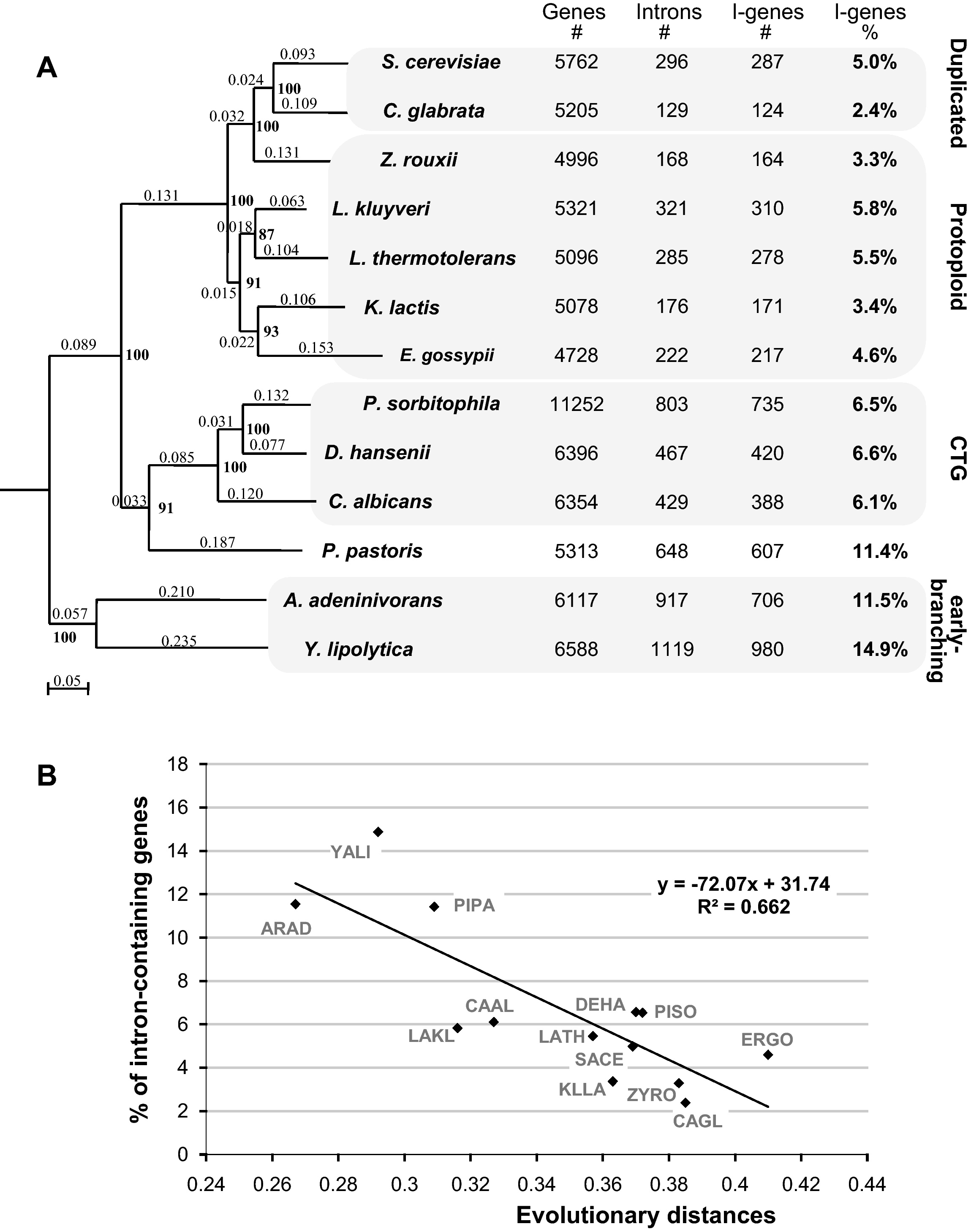
Correlation between evolutionary distances and intron loss. A. The phylogenetic tree is based on the concatenation of 84 protein alignments totalizing 6184 amino acid residues. Branch length is indicated next to each branch, and bootstrap values (in bold) next to each node. S. pombe was used as outgroup. The number of introns, intron-containing genes (I-genes) and the percentage of I-genes in each genome are indicated on the right of each species name. B. Relationship between evolutionary distances established from the tree in Fig. 1A and the percentage of I-genes in each genome. The black line corresponds to a linear tendency curve calculated from the data; its equation and the associated correlation coefficient (R2) are given on the graph. A Spearman test gives a pvalue of 3.8e-07 with a Rho of. −0.8155. Species names are as follows: S. cerevisiae (SACE), C. glabrata (CAGL), Z. rouxii (ZYRO), L. kluyveri (LAKL), L. thermotolerans (LATH), K. lactis (KLLA), E. gossypii (ERGO), P. sorbitophila (PISO), D. hansenii (DEHA), C. albicans (CAAL), P. pastoris (PIPA), A. adeninivorans (ARAD) and Y. lipolytica (YALI). Masquer
Correlation between evolutionary distances and intron loss. A. The phylogenetic tree is based on the concatenation of 84 protein alignments totalizing 6184 amino acid residues. Branch length is indicated next to each branch, and bootstrap values (in bold) next to ... Lire la suite
3.2 Conservation and variation of intron features
Based on previous analysis of splicing signal conservation [16,19], we considered the nucleotide environment of each intron/exon junction and of the BP (Fig. 2). In agreement with previous studies reporting that the information content in the 5'ss region (intron and exon) was the highest in hemiascomycetes and in the protozoan Cryptosporidium parvum [14,19], we observed that six nucleotides in the intronic 5'ss, seven nucleotides in the BP and 3 nucleotides in the 3'ss were highly conserved in almost all species. Contrary to most metazoans, the exonic part of the intron/exon junctions showed a lower level of information content which is coherent with an intron definition mechanism. The conservation of the three bases upstream and downstream of the introns appeared poorly conserved in the logo representation of Fig. 2. However, a systematic bias in upstream residues is detected for all species if we consider as preferentially conserved the nucleotides present in more than 40% of the nucleotides at this location, as previously reported [16]. The biased stretch of nucleotides is AAG for C. glabrata, E. gossypii, L. thermotolerans, C. albicans, P. pastoris and P. sorbitophila, AAN for K. lactis, L. kluyveri and Z. rouxii and ANG for D. hansenii, A. adeninivorans and Y. lipolytica. In contrast to what was previously predicted from a partial set of introns, no bias was detected in the first residues of the downstream exon [16].
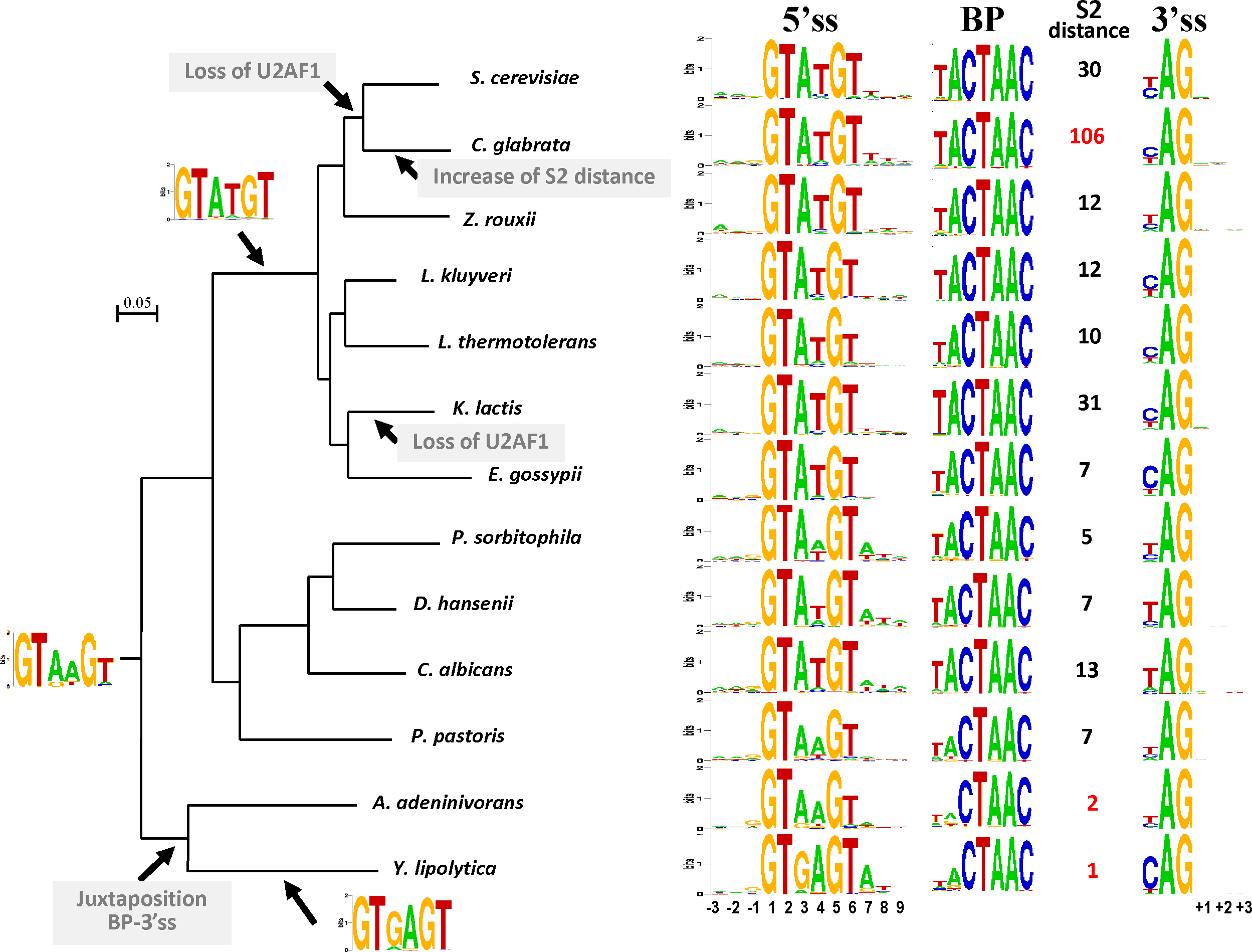
Evolution of intron structure and intron boundaries in budding yeasts. Conservation of 5'ss, BP and 3'ss are represented on the right of each species name as sequence logos. The three nucleotides upstream from the exon/intron junction (called −3, −2 and −1) and downstream of the 5'ss motif (called 7, 8 and 9), as well as three nucleotides downstream of the intron/exon junction (called +1, +2 and +3) are also represented. Distance between BP and 3'ss is indicated as S2 distance (in nt). The most extreme values, which correspond to C. glabrata, Y. lipolytica and A. adeninivorans S2 distances, are in red. The putative motif for ancestral 5'ss is represented on the phylogenetic tree next to the common ancestor node. Strong 5'ss motifs acquired by specific clade are indicated next to the corresponding branches as well as selected evolutionary events that had an impact on intron structure. Masquer
Evolution of intron structure and intron boundaries in budding yeasts. Conservation of 5'ss, BP and 3'ss are represented on the right of each species name as sequence logos. The three nucleotides upstream from the exon/intron junction (called −3, −2 and ... Lire la suite
Noticeable differences were observed among the consensus sequences in the different lineages of hemiascomycetes. Whereas GTATGT is the preponderant motif for 5'ss in the species deriving from the whole genome duplication [35], as well as in the protoploids [32], the motif the most used in A. adeninivorans and P.pastoris is GTAAGT and it is GTGAGT in Y. lipolytica. Species of the CTG clade, i.e. those which translate the CUG codons into serine instead of leucine, are intermediate. They use both GTATGT and GTAAGT with roughly equilibrated frequencies in P. sorbitophila and D. hansenii but with a pronounced predominance of GTATGT in C. albicans (Fig. 2). It is striking to observe that in P. sorbitophila, a recently formed spontaneous hybrid between two CTG species, the frequencies of GTAAGT and GTATGT are nearly equal (45% and 42%, respectively) but that this dual conservation does not correspond to the hybrid nature of the sequenced strain: each type of motif is equally distributed in the regions inherited from the two parental genomes. According to the 5'ss conservation in other ascomycetes, the most parsimonious ancestral motif may be GTRWGT with a preponderance of GTAAGT. Thus, some species may have preferentially retained the predominant ancestral motif (GTAAGT in A. adeninivorans and P. pastoris) or selected a less used motif becoming a strong 5'ss in the modern species: GTGAGT in Y. lipolytica and GTATGT in the common ancestor of duplicated species, and protoploids. We thus verified if the 5'ss differences were due to mutations in the U1 snRNA that base-pairs with the 5'ss during the early stage of spliceosome assembly (see [36] for a review on Spliceosome structure and function). The alignment of the U1 snRNA sequences showed that in all species the 9 first nucleotides that bind the 5'ss region were 100% identical (ATACTTACC) and matched perfectly (underlined characters) the GTAAGT ancestral motif. Thus, modifications in the 5'ss cannot be correlated with mutations in U1 snRNA. Similarly, the conservation of U6 snRNA that base-pairs at later stages with the 5'ss was investigated [37]. In S. cerevisiae, U6 residues 47-49 (ACA) pair with residues at position 4, 5 and 6 (UGU) of the 5'ss. In all hemiascomycetous species studied, the nucleotides equivalent to position 44 to 53 of S. cerevisiae snR6 that flank the pairing site are identical. Thus, in early-branching species, as in most eukaryotes in which the fourth nucleotide of intron is mainly an adenosine residue instead of a T in S. cerevisiae, the U6-5'ss pairing relies only on the two nucleotides G and U at positions 5 and 6 of the 5'ss. Once again, Y. lipolytica represents an exception with a high conservation of an A residue at position 7, i.e. the first nucleotide downstream of the 5'ss. This A residue allows to strengthen the base-pairing with U6 snRNA at position 46 (GUA base-pairs with UAC of U6 instead of AC only). The percentage of A residues at position 7 reaches 83% when the 5'ss is GTGAGT (784/946 cases) whereas it is only 50% with the 5'ss GTNTGT (9/18 cases). This finding suggests a covariation of 5'ss position 4 (A versus T) with position 7 (A versus not A) as previously reported for S. cerevisiae introns bearing a 5'ss GTACGT [38]. This covariation also exists in species of the CTG clade. For instance, in P. sorbitophila an A residue is present in 80% of the introns bearing GTAAGT (293/368 cases) whereas it is 49% in introns having GTATGT as 5'ss. In duplicated and protoploid species and in some CTG species, the situation is the same as in S. cerevisiae: the dominant U residue at position 4 that was selected across evolution increases the pairing and probably stabilizes the U6 interaction with 5'ss, having thus a functional consequence on spliceosome assembly.
The overall structure of intron is highly variable among yeasts. The mean length of introns varies from 106 nt in E. gossypii to 446 nt in C. glabrata. Most species show a bias towards short introns, and in some species, the range of intron size is quite broad. For instance, in Y. lipolytica, intron size varies from 41 to 3,478 bp with 16 introns larger than 1 kb [31]. Previous analyses revealed a bimodal distribution of intron size in S. cerevisiae [38,39] and suggested a size difference between introns of ribosomal protein genes (RPGs) and nonribosomal protein genes (NRPGs) in 7 hemiascomycetous species [16]. The intron size distribution was thus analyzed for the 13 species studied. A bimodal distribution was clearly observed for C. glabrata, K. lactis and E. gossypii, as reported in S. cerevisiae. This distribution correlates with the different distributions of intron size in RPGs and NRPGs (Fig. 3). In these four species, le mean size of RPG introns is more than twice the size of NRPG introns (Table 1), suggesting that these RPG introns may have undergone a specific evolution, some of them harboring snoRNAs ([10,38], see [10] for a list of snoRNAs in yeasts) and may require particular cis- or trans-acting factors for their splicing regulation as reported for S. cerevisiae [40]. In all other species, no bimodal distribution was clearly observed, however a difference still exists between the mean size of RPG and NRPG introns, RPG introns tending to be longer while those of NRPG are shorter. The absence of clear bimodal distribution is probably linked to the low proportion of introns in RPGs compared to the whole set of introns (from 3% to 16%) in these 9 species which may reflect their different dynamics of intron loss. For instance, Z. rouxii which contains as many introns as K. lactis has significantly lost more introns in RPGs, with only 26 remaining introns versus 46 in K. lactis (Fig. 3). More strikingly, A. adeninivorans which still contains 11.5% of intron-containing genes retained only 23 RPGs with at least one intron. More generally, the dynamics of intron loss is different from one species to another. For instance, the proportion of multi-intronic genes is highly variable among the studied yeast. In duplicated and protoploid species, multi-intronic genes carry at most two introns and their number ranges from 4 in Z. rouxii to 11 in L. kluyveri which reflects the situation in S. cerevisiae. However, only two families of these genes show a perfect conservation of both introns, the orthologues of the other genes bearing only one or no intron. In CTG and early-branching species, genes with three introns or more, up to seven in A. adeninivorans, have been predicted, multi-intronic-genes representing from 6.8% (P. pastoris) to 21.5% (A. adeninivorans) of all intron-containing genes. The distribution of introns in these genes is not conserved. For instance, the homologs of ARAD1D24024 g which bears 7 introns have only 3 introns in Y. lipolytica and none in P. sorbitophila and D. hansenii.
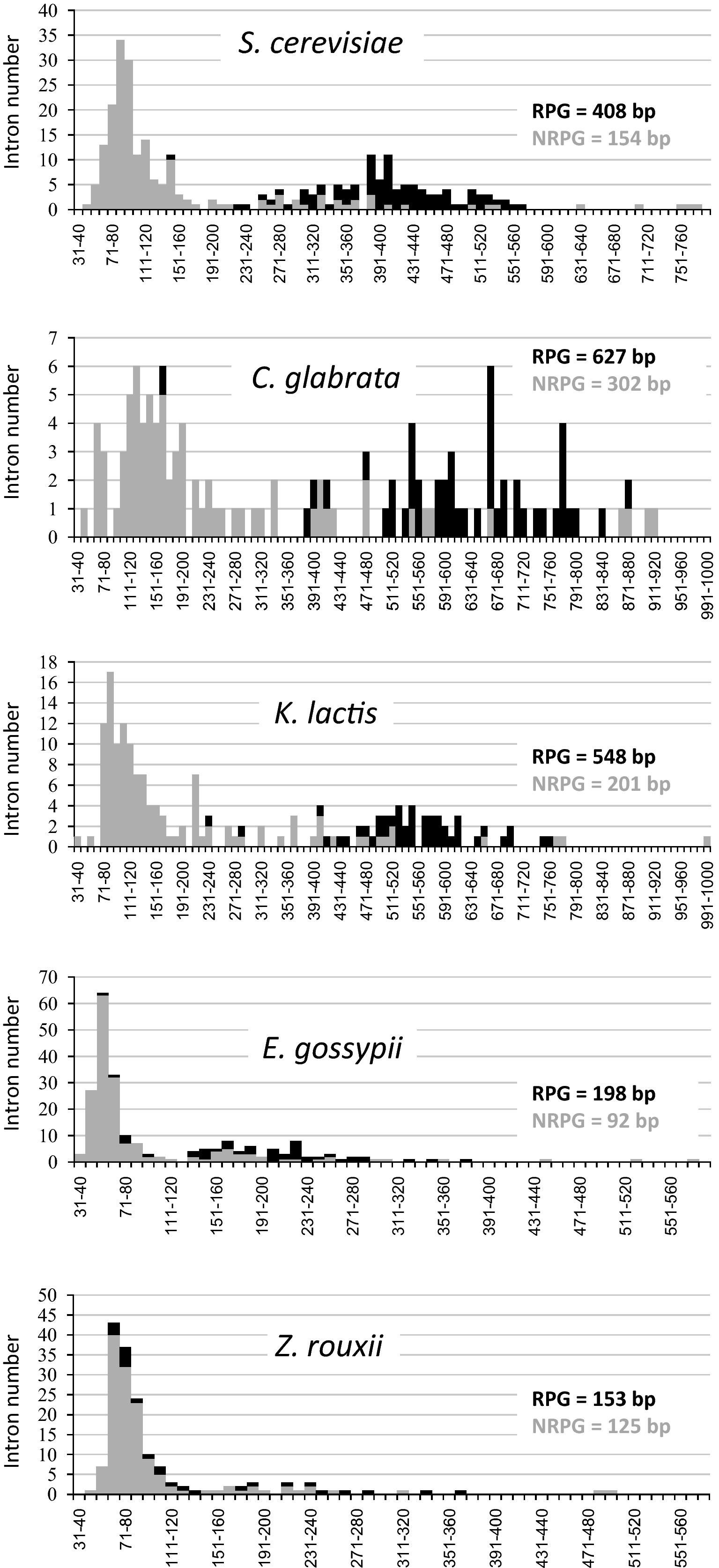
Distribution of intron size in ribosomal protein genes (RPGs) and non-ribosomal protein genes (NRPGs). Intron size of RPGs (in black) and NRPGs (in grey) are plotted on this cumulative histogram with size windows of 10 nt from 31 to 580, 790 or 1000 nt, depending on the intron size of each species. Four species with a clear bimodal distribution are represented: S. cerevisiae, C. glabrata, K. lactis and E. gossypii, as well as an example of species without bimodal distribution: Z. rouxii.
Number and size of introns in genes encoding ribosomal proteins (RPG) or non ribosomal proteins (NRPG).
| Species | Ribosomal Protein Genes | Non Ribosomal Protein genes | I-RPG/All Introns % | |||||
| Genes # | Introns # | I-genes # | Mean I-size nt | Introns # | I-genes # | Mean I-size nt | ||
| S. cerevisiae | 137 | 91 | 89 | 408 | 205 | 198 | 154 | 31 |
| C. glabrata | 85 | 46 | 46 | 627 | 83 | 78 | 302 | 36 |
| Z. rouxii | 82 | 26 | 25 | 153 | 142 | 139 | 125 | 15 |
| L. kluyveri | 81 | 48 | 47 | 406 | 273 | 263 | 123 | 15 |
| L. thermotolerans | 82 | 46 | 45 | 306 | 238 | 232 | 117 | 16 |
| K. lactis | 82 | 46 | 44 | 548 | 129 | 126 | 201 | 26 |
| E. gossypii | 80 | 46 | 44 | 198 | 176 | 173 | 92 | 21 |
| P. sorbitophila | 172 | 92 | 90 | 325 | 711 | 645 | 119 | 11 |
| D. hansenii | 88 | 50 | 49 | 341 | 417 | 371 | 108 | 11 |
| C. albicans | 84 | 54 | 53 | 388 | 375 | 335 | 152 | 13 |
| A. adeninivorans | 78 | 27 | 23 | 141 | 890 | 683 | 86 | 3 |
| Y. lipolytica | 81 | 56 | 56 | 308 | 1063 | 924 | 271 | 5 |
Considering intron structure evolution, the most striking size difference lies in the position of the BP relative to 5'ss and 3'ss. According to the distance between BP and 3'ss, also called S2 distance, fungal introns have been divided into two classes: the 3′S class regroups introns with a S2 distance ranging from 5 to 15 nt and 3′L introns are characterized by longer S2 distances with a uridine-rich segment. Differences in the mechanistic for spliceosome assembly are associated with each of these two classes. In the 3′S introns, the two conserved residues AG at the 3′ end of introns base-pair with the snRNA U1 and are required for lariat formation whereas this interaction is not necessary for 3′L introns [41,37]. Whereas most S. pombe introns belong to the first class, those of S. cerevisiae are mainly 3′L introns. In all hemiascomycetous species studied here the S2 distance usually ranges from 5 to 13 nt, thus characteristic of the 3′S class, but some exceptions exist. In Y. lipolytica and A. adeninivorans, the BP and 3'ss form a 11 or 12-nt consensus sequence in which the S2 distance corresponds to a single nucleotide in Y. lipolytica and to two nucleotides in A. adeninivorans. This juxtaposition may reflect a simplified splicing mechanism as proposed for Trichomonas vaginalis and Giardia intestinalis, two deep-branching protists in which introns have a highly conserved 12-nt 3'ss/BP motif [42]. However, Schwartz et al. [19] proposed an alternative hypothesis based on a partial function loss of U2AF2 in Y. lipolytica. This splicing factor may have conserved its ability to bind U2AF1 and SF1 homologs, two main splicing factors that tether the BP and 3'ss of introns, but may have lost its ability to bind the PPT. Thus, Y. lipolytica U2AF2 protein may only serve as a bridge between U2AF1 and SF1 and thereby could constrain the distance between BP and 3'ss. The second type of unusual S2 distance was found in C. glabrata and to a lesser extent in S. cerevisiae and K. lactis. In all these species, the S2 distance has tremendously increased and reached a median value of 106 bp in C. glabrata, which enable to classify them as 3′L introns. However, in the absence of experimental data, it is difficult to know if C. glabrata and K. lactis follow the same steps as S. cerevisiae during the early phases of splicing complex formation, and for instance if they are also AG-independent. Thus, as in the case of Y. lipolytica, we tried to find a correlation common to these three species between S2 distance and the presence or the functionality of splicing factors, especially U2AF1, U2AF2 and SF1 (Fig. 2). Homologs of U2AF2 (also called MUD2 in S. cerevisiae; [20]) and SF1 were identified in the three species (Table 2). In contrast, no homolog was found in the three species for U2AF1. Whereas U2AF2/MUD2 is poorly conserved across the eukaryotic tree, U2AF1 homologs are highly conserved. Käufer and Potashkin [43] reported 75% of similarity between S. pombe U2AF1 (also called U2AF25) and human U2AF1. Thus, this protein has probably been lost in the common ancestor of S. cerevisiae and C. glabrata. In K. lactis, the loss was lineage specific. The fact that U2AF1 has been lost in these two different lineages suggests a correlation with the increase of the S2 distance. Human U2AF1 (U2AF35) is known to interact with the AG dinucleotide at the 3'ss [44]. The lack of binding to 3'ss in S. cerevisiae may be compensated by the efficient interaction between SF1 and the strong consensus of BP (UACUAAC) or by the interaction between U2AF2 and the PPT [45]. Moreover, in vivo assays showed that the presence of a uridine-rich motif in S. cerevisiae enables distal PyAG (> 30 bp) to compete as a 3'ss [46]. These findings could justify the decrease of constraints on the S2 distance.
Conservation of splicing factors binding to 3’ end of introns.
| Species | MUD2/U2AF2 | U2AF1 | MSL5/SF1 |
| S. cerevisiae | + | Lost | + |
| C. glabrata | + | Lost | + |
| Z. rouxii | + | + | + |
| L. kluyveri | + | + | + |
| L. thermotolerans | + | + | + |
| K. lactis | + | Lost | + |
| E. gossypii | + | + | + |
| P. sorbitophila | + | + | + |
| D. hansenii | + | + | + |
| C. albicans | + | + | + |
| P. pastoris | + | + | + |
| A. adeninivorans | + | + | + |
| Y. lipolytica | + | + | + |
| Génolevures family | GL3C2997 | GL3R3615 | GL3R1217 |
3.3 Genosplicing database
The Genosplicing website was developed and dedicated to spliceosomal introns of yeasts. The intron pattern is defined as the overall characteristics of the entire intronome of a given species. It includes frequencies and statistics for intron size, S1 and S2 distances, as well as consensus motifs for 5'ss, BP and 3'ss either expressed as frequencies and represented as sequence logos. Genosplicing provides intron patterns of fully sequenced genomes with easily extractable data on spliceosomal introns. For some species, the entire set of intron sequences and CDS or proteins of intron-containing genes is downloadable. A search facility enables to query the database to know if particular genes contain spliceosomal introns. We hope that this website will facilitate gene model prediction in further sequenced genomes.
3.4 Alternative splicing in yeasts
Alternative splicing (AS) is currently thought to be a major source for increasing the complexity of transcriptomes and proteomes, and to lead in some cases, to genetic or malignant diseases in mammals especially in human where intron density is high [47–50]. Despite the fact that yeasts have intron-poor genomes, some examples of AS have been reported. Few of them result in the production of different proteins, as reported in Schizosaccharomyces pombe or S. cerevisiae [51,52]. More often, alternative transcripts have been predicted without evidence for multiple functional proteins [15,53,54]. With the development of global transcriptomic approaches (tiling arrays or RNAseq), many cases of AS have been identified in various yeasts, increasing unexpectedly the complexity of their transcriptomes [55–58] but not of the proteome as often they result in nonsense-containing mRNAs due to intron retention [31]. These noncoding alternative transcripts were thought to be largely non functional. However, in some cases, intron retention occurred at a specific physiological state of the cells, such as meiosis [23,59,60] or under specific growth conditions, such as amino-acid starvation [61], suggesting that AS may be regulated. Other examples of regulated splicing involving complex mechanisms were reported in S. cerevisiae such as those of RPL30 [40] and YRA1 [62,63], in which the protein inhibits the splicing of its own pre-mRNA.
4 Concluding remarks and perspectives
About thirty hemiascomycetous genomes have been sequenced since that of S. cerevisiae [64] and it is now possible to compare the full in silico predicted intronome of divergent yeast species. We have shown here that all the hemiascomycetes are justly considered as intron poor genomes and that their intron boundaries and architecture have undergone numerous evolutionary events, some of them leading to lineages specific features. Such analyses should now be extended to genomes from unexplored clades such as those that mainly diverged early in the phylogenetic tree of Hemiascomycetes. With the new sequencing technologies, the number of fully sequenced genomes will dramatically increase in the near future and we hope that interest for hemiascomycetous transcriptomes will experience the same enthusiasm.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We would like to thank all our colleagues from the Génolevures consortium for helpful discussions and for the access to the two genomes not publicly available, P. sorbitophila and A. adeninivorans. This work was financially supported by the GDR CNRS 2354 “Génolevures-3” and the ANR “Genarise” (ANR-05-BLAN-0331).
Vous devez vous connecter pour continuer.
S'authentifier