

1 Introduction
The Saccharomyces genus [1,2] currently contains eight species, several of which play a major role in food or beverage fermentations (Fig. 1). Saccharomyces cerevisiae, Saccharomyces bayanus and Saccharomyces pastorianus are associated with anthropic environments, whereas Saccharomyces paradoxus, Saccharomyces kudriavzevii, Saccharomyces cariocanus, Saccharomyces mikatae and the recently described Saccharomyces arboricolus are mostly isolated from natural environments. Most of the yeast strains used for alcoholic fermentation are now recognized as S. cerevisiae. The cryotolerant species S. bayanus var. uvarum (or S. uvarum, [3]) may also be used for alcoholic fermentation, particularly in winemaking at low temperature [4] or cider production [5,6]. Some S. paradoxus strains have been isolated from vineyards, but they seem to make little contribution to wine fermentation [7]. S. pastorianus is a hybrid species used for the production of lager beer (bottom fermentation), whereas the ale yeasts involved in top fermentation mostly belong to the species S. cerevisiae. Saccharomyces species can mate with each other and form viable F1 hybrids that can grow asexually, but are sterile [8]. In the last decade, a growing number of natural inter- and intraspecific hybrids have been identified, predominantly among domesticated yeasts [7,9].
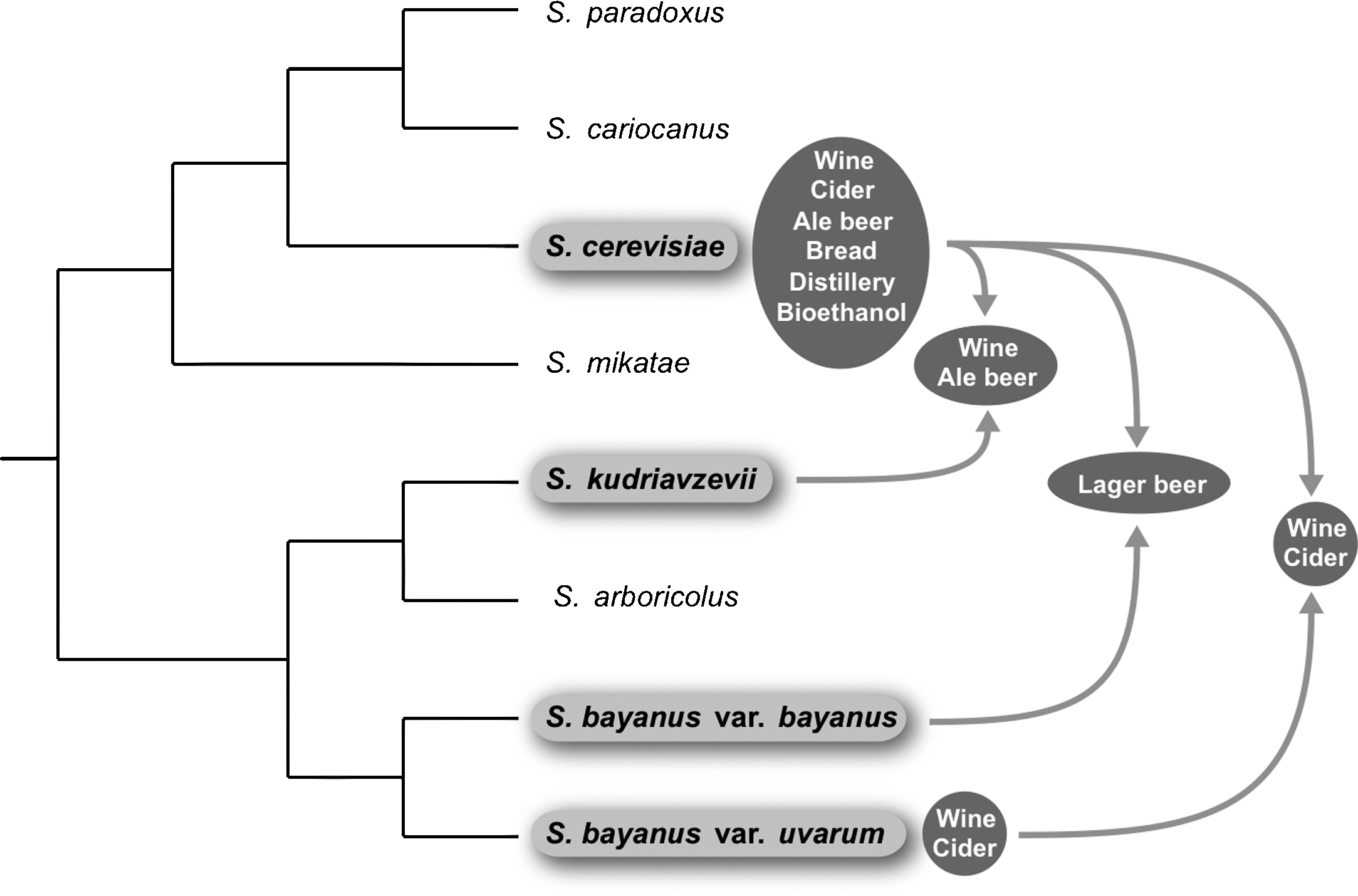
Schematic diagram of the phylogenetic relationships between the Saccharomyces species and their industrial specialization. The tree is adapted from Naumov et al. [75]. The species involved in industrial processes and/or in hybrids are boxed in light grey. The products of industrial processes involving the hybrids and non-hybrids are boxed in dark grey. The arrows correspond to hybrids. Only the contributors of these hybrids are shown. Those of the lager beer S. pastorianus are S. cerevisiae and S. bayanus var. bayanus.
2 Evolutionary history and genetics of fermentative Saccharomyces cerevisiae
S. cerevisiae is of considerable importance for the baking, brewing, winemaking and distillation industries. This yeast has been exploited by humans for millennia, for the fermentation and preservation of beverages and food. It is thought that beverage and bread fermentation technologies expanded outwards from Asia, Mesopotamia and Egypt to the rest of the world. The earliest evidence for the production of fermented beverages has been dated to 7000 BC in China [10,11]. Several recent studies, based on Multilocus sequence typing (MLST), multilocus microsatellite analysis, genome sequencing and whole-genome tiling microarrays, have revealed the impact of domestication on yeast genetic structure [12–19]. Remarkably, most vineyard isolates form a group clearly separated from other technological groups, providing evidence for a single domestication event followed by the human-associated migration of wine yeasts throughout the world. The strains used for sake production also cluster independently, consistent with a second, independent domestication event.
The S. cerevisiae strains involved in fermentative processes have unique genetic and phenotypic features. Wine yeasts are predominantly diploid [17,20], homothallic [21] and mostly heterozygous (65%), with variable sporulation ability [22]. No evidence for aneuploidy has been detected by comparative genomic hybridisation (CGH) [23,24]. However, flor yeasts (film-forming yeasts involved in sherry production), despite their phylogenetically close relationship to other wine yeasts [17], have a more complex genetic makeup. Both genetic and CGH studies have suggested that aneuploidy is common in these yeasts [25–27]. Bread and brewing yeast strains have ploidy levels greater than 2n. Ale beer strains have four alleles at several loci, suggesting partial (aneuploidization) or whole-genome duplication (polyploidization) [17]. Recent studies have shown that most bread strains are tetraploids that may have been generated by the fusion of two nonreduced meiospores from two S. cerevisiae strains (one ale beer strain and one wine strain) [17,28].
3 Genome variation and adaptation mechanisms
Over time, the use of yeasts by humans for fermentation has led to the selection of specialized strains for baking, brewing and winemaking. These specialized S. cerevisiae strains are not readily interchangeable and display specific phenotypic traits, some of which are clearly shared by strains from the same ecological niche. One of the key challenges in comparative genomics is tracking the footprints of evolution in the genomes of these strains, which have been shaped by various selective forces. The knowledge accumulated to date, particularly for wine yeast genomes, indicates that the genomic diversity of these genomes has been shaped by many different mechanisms (for reviews see [29] and [30]). We will focus here on the most recent advances in our understanding of such genomic mechanisms.
3.1 Gross chromosomal rearrangements (GCR)
It was shown very early that wine yeasts display a high level of chromosomal length polymorphism [31,32] as a result of GCR events, including translocations, deletions and amplifications of chromosomal regions [33,34]. These events are mediated by ectopic recombination between repeated Ty sequences or duplicated genes [35–37]. Comparative genome hybridisation (CGH) recently highlighted the role of retrotransposon mobility in generating variability in wine strain genomes [23]. The genome sequence of the EC1118 wine yeast has recently been shown to contain several chromosomal translocations and insertions of blocks of DNA not present in the genome of the reference strain S288C. Many of these translocations and insertions are located in peripheral regions of the chromosomes (unpublished data), as also reported for the clinical isolate YJM789 [38], consistent with the view that peripheral regions are highly plastic and free to undergo ectopic recombination. In most cases, it remains unclear whether these rearrangements contribute to yeast fitness. A reciprocal translocation between chromosomes VIII and XV, that is widespread in wine yeasts, increases the expression of SSU1, which encodes a plasma membrane protein involved in sulfite anion extrusion, thereby conferring sulfite resistance [39,40]. This translocation has been selected by the extensive use of sulfite in winemaking, and constitutes a clear example of the contribution of GCR to adaptation.
Flor yeasts have also been reported to contain many GCR. The flor fermentation process is associated with selective and mutagenic conditions. The exposure of yeasts to high levels of acetaldehyde and alcohol may induce double strand breaks, the processing of which may favour GCR [26].
3.2 Copy number variations (CNV)
Gene amplification is a common mechanism of adaptation used by various organisms to cope with stressful environments or to increase resistance to various toxic agents. Microarray-based whole-genome hybridisation studies of various S. cerevisiae strains [19,41–43] have uncovered a recurrent pattern of CNV, suggesting a role for repetitive DNA sequences in structural genome diversification and adaptation to specific environments. CGH analyses of wine yeast strains have shown both the duplication and deletion of subtelomeric genes [23,24]. Many of the differences between strains seem to concern transporter genes and genes involved in drug responses [24]. CNV has also been observed in telomeric or subtelomeric chromosomal regions of the S. cerevisiae strains used in biofuel production. CGH analysis of five industrially important fuel ethanol S. cerevisiae strains revealed amplifications of the telomeric SNO and SNZ genes, which are involved in the biosynthesis of vitamins B6 (pyridoxine) and B1 (thiamin). It has been suggested that this increase in the copy number of these genes enables these strains to grow more efficiently in sugar cane juice [42].
3.3 Sequence polymorphisms
Recent genomic surveys have estimated the overall DNA divergence between strains at 1 to 1.4 substitution per kilobase for vineyard isolates and five to six substitutions per kilobase between wine strains and other S. cerevisiae strains [15,18,19]. In a few cases, sequence polymorphisms have been shown to influence phenotypes of industrial relevance. For example, flor yeast strains have acquired two mutations in FLO11, a key gene for the formation of velum encoding a cell wall mucin. These mutations, in the promoter and the coding region of FLO11, increase the expression of this gene and the ability of cells to adhere to each other, respectively [26,44]. Another example was reported by Guillaume et al. [45], who identified a mutated HXT3 allele in some wine yeast strains that conferred an enhanced capacity to ferment fructose.
3.4 Introgressions
The acquisition of genes by DNA transfer has long been regarded as rare in yeasts. However, the genome of the S. cerevisiae wine yeast EC1118 was found to contain unexpectedly large chromosomal segments acquired through independent HGT events from different donors, including non-Saccharomyces species [46]. Several of these regions are widespread among wine yeast strains. These introgressions carry 34 novel genes with functions mostly related to nitrogen and carbon metabolism, suggesting a potential role in the adaptation of strains to the winemaking environment. The functions of some of these genes have been determined, although most remain unknown. One of these genes is homologous to S. pastorianus FSY1 and encodes a high-affinity fructose symporter, fulfilling a function not found in S. cerevisiae (Fig. 2). This transporter may confer an advantage on yeast cells at the end of wine fermentation, when most of the remaining sugar is in the form of fructose [47]. Two tandem duplicated genes encoding proteins resembling nitrogen permeases [46] were recently shown to encode oligopeptide transporters. These genes belong to a new family of fungal oligopeptide transporters (FOT) identified by functional metatranscriptomic analyses of eurkaryotic soil microbial communities [48]. The presence of these carriers makes it possible for yeast to transport a much broader range of oligopeptides than are usually transported by the S. cerevisiae carriers Ptr2p and Dal5p (Fig. 2) and likely contributes to the metabolic diversity regarding the utilization of di/tripeptides [49]. As nitrogen levels are usually very low at the end of fermentation, the use of dipeptides may provide wine yeasts containing Fot transporters with a nutritional advantage at the end of alcoholic fermentation. FSY1 and FOT genes are located on a 65-kb DNA fragment found in the subtelomeric region of the right end of chromosome XV of many wine yeast strains. The contributor of this region may be a species closely related to the Saccharomyces genus [46, Fig. 2]. Interestingly, this DNA fragment also carries the XDH1 gene (EC1118_1D0_6623g, Fig. 2) that encodes a putative xylitol dehydrogenase recently identified by bulk segregant analysis in xylose-utilizing wine yeasts [50].
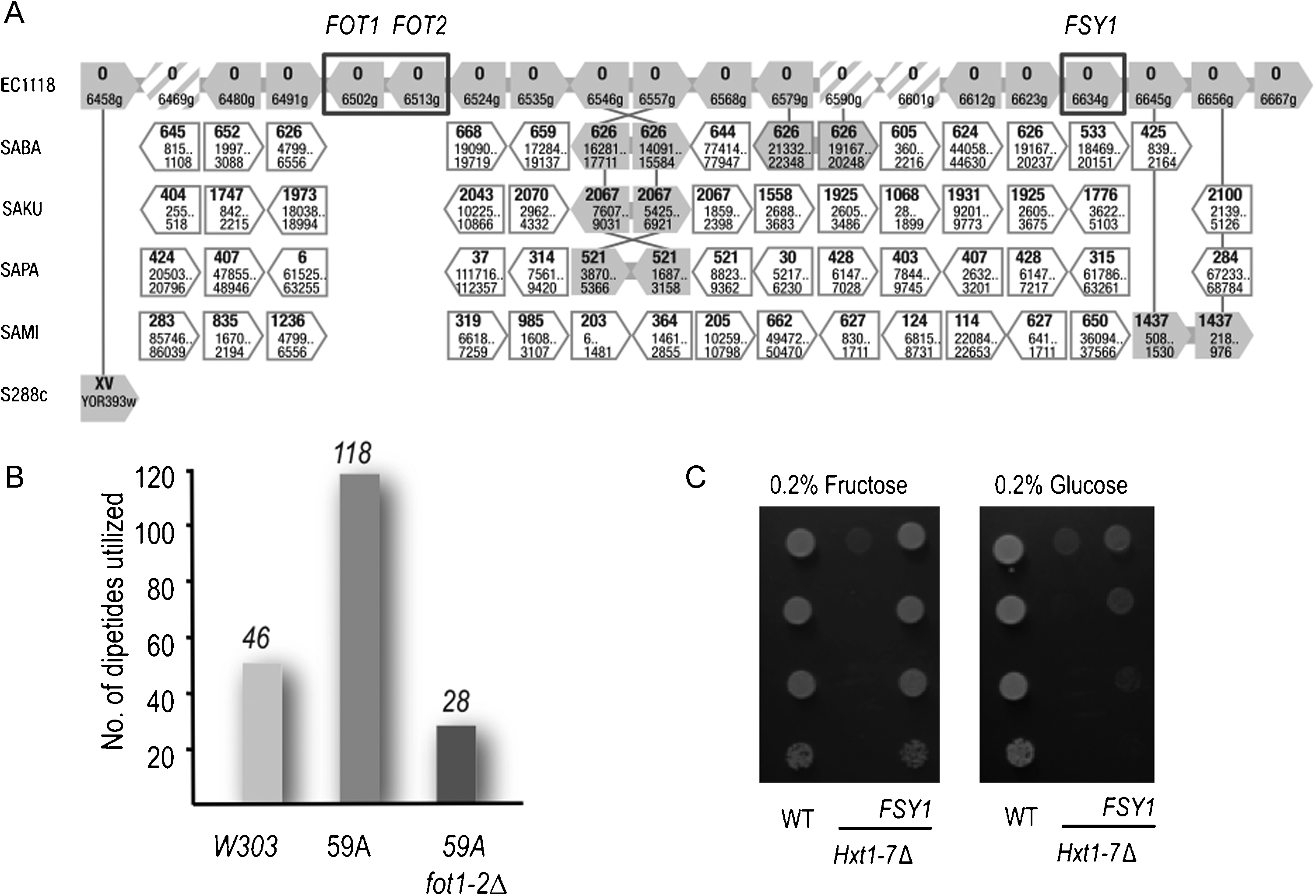
Function of genes acquired by horizontal transfer in wine yeast genomes. A. Synteny of the genes in EC1118 region C (subtelomeric end of chromosome XV) and in various Saccharomyces species, adapted from Novo et al. [46]. Filled arrows: genes displaying synteny with those in EC1118. Hatched arrows: pseudogenes. S. bayanus (SABA), S. kudriavzevii (SAKU), S. paradoxus (SAPA) and S. mikatae (SAMI). B. Number of oligopeptides transported by the S. cerevisiae strains W303A (laboratory strain without region C), the haploid derivative of EC1118 (59A) and the 59A fot1-2Δ strains. C. Growth of V5 hxt1-7Δ expressing the FSY1_EC gene on synthetic medium with different carbon sources.
The donor of one of the other introgressions found in wine yeast genomes is a wine contaminant (Zygosaccharomyces bailii), suggesting that these events may be facilitated by ecological proximity [46]. This region is widespread in wine yeasts (it was detected in more than half of 52 wine strains) and displays a low level of diversity, confirming the recent origin of this event, after the start of wine strain expansion [46,51]. It was also recently shown that this introgression is found in multiple copies in wine yeast genomes, at various chromosomal positions. The organization of these different forms and the presence of an autonomously replicating sequence functional in S. cerevisiae suggest a circle-based expansion mechanism, generating interchromosomal amplifications mediated by nonhomologous recombination [51]. The uptake, maintenance and expansion of these foreign genes in the genome of wine yeast strains suggests that they contribute in some way to increasing the evolutionary fitness of wine yeasts.
Evidence for the existence of novel genes has also been found in the genomes of other S. cerevisiae strains [18], raising questions about the frequency of such events in this species.
4 Saccharomyces hybrids
Industrial Saccharomyces yeasts are unusual in being mostly interspecific hybrids. It is generally accepted that the hybrid nature of their genomes is an advantage as it brings together characteristics from each of the parental stains. Their characteristics as hybrids, including the species giving rise to them and the complexity of their genomes, vary with specialization and industrial environment (Fig. 1). All were generated with S. cerevisiae as the fermentative parent. Saccharomyces interspecific hybrids have been described in two recent reviews [7,9]. We review here the most recent insights into Saccharomyces hybrids.
The most complex hybrids are the only ones that have been classified as a species, S. pastorianus (= Saccharomyces carlsbergensis). These strains are responsible for bottom fermentation in lager beer production. Their hybrid nature was discovered many years ago [52,53], but it has only recently been shown, by molecular methods, that the emergence of these strains from two main contributors, S. cerevisiae and S. bayanus var. bayanus, was recent [54,55]. It has even been suggested that some S. pastorianus strains may be triple hybrids between these two species and another as yet unidentified species [55]. Brewing yeast genomes are allopolyploid, with major rearrangements and gains and losses of chromosomes or parts of chromosomes; they thus display tremendous intraspecific variability, as reviewed elsewhere [53]. CGH analyses confirmed these observations [56,57]. One brewing yeast hybrid, Weihenstephan 34/70, has been sequenced [58]. This sequence, the first obtained for a yeast hybrid, confirmed what was known about S. pastorianus: the allopolyploid nature of its genome and the identification of S. bayanus var. bayanus, rather than var. uvarum, and S. cerevisiae as the parental species. An analysis of rDNA showed intraspecific variability, with the rDNA units originating from S. bayanus var. bayanus tending to be lost. Synteny analysis indicated that post-hybridisation events had occurred. Telomeres were shown to carry genes non-orthologous to those of S. cerevisiae, adding to the originality of brewers’ yeasts, because the telomeres are known to carry genes involved in beer making. Maltose and maltotriose transporter genes and genes involved in sulfite production were also present at higher copy numbers in the sequenced brewing yeast genome, consistent with the physiological properties of the brewing hybrids.
One study [57] suggested that brewing yeasts have two origins. In the first group, which includes the sequenced strain, Weihenstephan 34/70, each of the parents has made an equal contribution (Group 1). In the second group, there is an imbalance, with S. cerevisiae contributing more material than the other parent (Group 2). This is also consistent with the difference in Tys distribution observed in these strains [59]. It has been suggested that the S. cerevisiae parent of the brewing yeast S. pastorianus was an ale strain (i.e. an S. cerevisiae top-fermenting strain), consistent with the ale and lager (bottom-fermenting) strains having the same niche. Interestingly, a comparison of the constitution of the genomes of the two lager groups led to the suggestion [57] that they originated through two independent events: interspecific hybridisation between two spores of S. cerevisiae and S. bayanus var. bayanus and fusion between a diploid S. cerevisiae and an haploid S. bayanus var. bayanus.
Ale strains were thought to belong to S. cerevisiae, but some ale strains have also been shown to be S. cerevisiae/S. kudriavzevii hybrids. RFLP analysis of PCR-amplified nuclear markers revealed that many of these strains were diploid, but with a complex genome constitution [60]. Complex genome constitutions can lead to genome instability: CGH has shown this to be the case for S. pastorianus, in which the fermentation of a high-gravity wort and high temperatures have been shown to induce major chromosomal changes [61].
Finally, hybridisation, which was long considered to occur only in brewers’ yeast strains, has proved common throughout Saccharomyces, with the discovery of wine and cider yeast hybrids between S. cerevisiae and S. bayanus var. uvarum, and of S. cerevisiae/S. bayanus var. uvarum/S. kudriavzevii and S. cerevisiae/S. kudriavzevii hybrids [62–64]. In their RFLP analysis of PCR-amplified nuclear and mitochondrial markers, Gonzalez et al. [65] identified such hybrids among strains originating from Switzerland. Similar hybrids were observed among Austrian wine strains [66]. It has been suggested that, after a single hybridisation step, the hybrid genome undergoes extensive chromosomal rearrangement, including chromosome losses and the generation of chimeric chromosomes by nonreciprocal recombination between homeologous chromosomes [67]. The advantages of hybrids over non-hybrids in wine fermentations have been discussed elsewhere [68–72]. The industrial fuel ethanol yeast strains studied to date are not hybrid. The recent use of these strains is consistent with the selection of high-performance hybrids requiring a certain amount of time.
5 Future developments
Our understanding of the genomes of fermentative Saccharomyces has now entered a golden age, with the widespread availability of high-throughput sequencing techniques. In recent years, a growing number of natural inter-specific hybrids of Saccharomyces species associated with human activity have been identified. This and the finding in wine yeast genomes of introgressions from distantly related yeasts sharing the same habitat suggest that fermentative Saccharomyces strains are able to share genetic material, thereby generating diversity. Massive amounts of genomic data are currently being produced and it seems likely that the genome sequences of several yeast hybrids not well characterized to date will become available in the near future. In addition, the genomic exploration of a larger number of strains from complex ecosystems should improve estimates of the frequency of gene transfer events and facilitate identification of the underlying mechanisms.
Yeast is one of the most widely used model organisms in genetics and is now also becoming an exciting model system in ecology and evolutionary studies. In addition to providing new insight into evolutionary mechanisms, studies of biodiversity should help us to decipher genotype-phenotype relationships. The sequencing of many yeast strains should facilitate genomic population studies and lead to the detection of more signatures of selection. Genomic data will also clearly provide us with a great opportunity to bridge the gap between genome variations and industrial traits. Quantitative genetics approaches should help to identify polymorphisms affecting the technological or sensorial properties of yeasts [73,74]. Information about the genome-wide distribution of SNPs should shortly become available for many fermentative strains, providing a considerable impetus to research in this field.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
This work was supported by INRA. This work has received funding from the European Community's Seventh Framework Programme (FP7, 2007–2013), Research Infrastructures action, under the grant agreement No. FP7-228310 (EMbaRC project).