

1 Introduction
Cystic fibrosis (CF) is the most frequent autosomal recessive genetic rare disease in the western world [1,2]. Mutations of the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) gene are the cause of CF. Since the discovery of the gene in 1989 [3,4], more than 2000 mutations have been identified. The c.1521_1523delCTT (p.Phe508del) mutation is present in two thirds of the CFTR mutated alleles, whereas the other mutations are private or limited to a small number of individuals [5].
Six classes of mutations have been established [6–8] to categorize the CFTR anomalies according to their impacts on mRNA synthesis (class I), splicing (class V), on protein processing (class II), stability (class VI) or functionality (class III and IV). Therefore, the classification of each newly identified mutation helps in predicting potential genotype–phenotype correlation and in tailoring new drug strategies.
Among the CFTR identified mutations, 40% are missense and lead to amino acid substitutions in one out of the five CFTR domains. CFTR protein is a multi-domain chloride channel composed of two Membrane Spanning Domains (MSD1 and MSD2), two Nucleotide Binding Domains (NBD1 and NBD2), and a regulatory domain [3]. The acquisition of a functional channel requires the coordinated folding and assembly of the CFTR domains [9,10]. Depending on the substitution localization in the CFTR domains, a missense mutation may dramatically alter the proper folding and/or functionality and induce a CF disease or CF-related disorder (CFTR-RD).
In this paper, the c.965T > C (p.Val322Ala) CFTR rare missense mutation is studied in cellulo after its identification in a French family (Brittany) in 1995 [11]. This exon 8 mutation induces a substitution of the valine322 into alanine at the fifth helix of the MSD1.
Our in cellulo studies showed that the c.965T > C mutation has no effect on the processing, localization and functionality of the p.Val322Ala CFTR protein in the tested cultured cell lines.
2 Material and methods
2.1 Construction of CFTR-mutated cDNA
CFTR constructions were created in the pTCF expression plasmid (provided by Dr. Pascale Fanen) with the Gene tailor site-directed mutagenesis kit (Invitrogen) using specific primers namely; (VF), 5’CTTTGTGGTGTTTTTATCTG[C]GCTTCCCTAT3’ and (VR) 5’CTTCTTCTCAGGGTTCTTTGTGGTGTTTTTATCTG3’. All mutants were controlled by sequencing using the ABI PRISM Big Dye Terminator™ Cycle Sequencing Ready Reaction Kit (Applied Biosystems) and specific primers (5’GATTGAAAACTTAAGACAAACAG3’ and 5’CATGGCGGTCACTCGGC3’). Reactions were run on an ABI PRISM™ 3100 automatic sequencer (Applied Biosystems).
2.2 Cells and transient transfections
HeLa cells were cultured in DMEM medium with Glutamax-I (Life Technologies) at 37 °C in the presence of 5% of CO2. Each medium was completed with 10% fetal bovine serum (Gibco), 100 units/mL of penicillin and 100 μg/mL of streptomycin. Three independent transfections were performed for immunolocalization, western blot (WB), and functionality tests.
2.3 Western blotting
Twenty-four hours after transfection, HeLa cells were harvested and resuspended in a lysis solution: 10 mM Tris-HCl (pH7.5), 1% Nonidet P-40, 0.5% deoxycholate supplemented with a protease inhibitor cocktail (Roche Applied Science) and 2 mM AEBSF. Proteins were quantified using BCA Protein Assay Kit (Sigma-Aldrich). 50 μg of protein were mixed with an equal volume of 2× Laemmli sample buffer (Sigma-Aldrich). Proteins were analysed on a 5% SDS-PAGE and transferred to nitrocellulose membranes using an iBlot system (Invitrogen) (20 V, 7 min). Blocked membranes with 5% non-fat dry milk were incubated with primary antibody against CFTR (clone M3A7; Chemicon) (1/2000 in 1X PBS) overnight. Membranes were then washed three times with TBST and incubated with secondary antibody conjugated to Alexa Fluor® 555 (1:100 in PBS) for 6 h. The blots were scanned with a Typhoon imager (GE Healthcare) using an excitation laser (532 nm) and a 580-nm band-pass filter (580 BP 30). The bands were quantified by densitometry using Scion Image software, and the CFTR maturation status was estimated by the band C/bands (B + C) ratio.
2.4 Cellular localization
HeLa and HEK 293 cells grown on glass were fixed in 4% paraformaldehyde and permeabilized as described previously [12]. Coverslips were incubated with human anti-CFTR MAB25031 (Invitrogen) (1/400 in 1X PBS) for 1 h at 4 °C followed by the secondary antibody, conjugated to Alexa Fluor® 555 (Invitrogen) (1/800 in 1X PBS) (40 min). Negative controls were also prepared in parallel for the comparison of positive treatment. Images were obtained using a confocal microscope (Olympus FV100) equipped with a multi-line Argon laser (457 nm, 488 nm, 515 nm) to visualize GFP, and with a HeNe green laser (543 nm) to visualize CFTR.
2.5 Functional tests by iodide efflux studies
CFTR chloride channel activity was evaluated on transfected HeLa cells treated with radioactive iodide (125I) as previously described [13,14]. The 125I efflux was realized by the MultiPROBE®IIex robotic liquid handling system (PerkinElmer Life Sciences, France) and measured by the Packard CobraTMII gamma counter (PerkinElmer Life Sciences, France). Statistical analysis was obtained using GraphPad Software. To compare sets of data, we used Student's t-test. Values were considered as statistically significant for P < 0.05, ns: non-significant difference. In the functional tests, the pTCF-WT is used as a positive control and pTCF-c.1392G > T (p.Lys464Asn) is used as a negative control instead of the pTCF-c.1521_1523delCTT, as it has been demonstrated that the c.1392G > T mutation is a class-II mutation [12].
3 Results
3.1 Proper folding and processing of the p.Val322Ala CFTR protein in the ER
To evaluate the effect of the c.965T > C mutation (p.Val322Ala) on the protein maturation and processing, WB analysis was used on HeLa cells that had been transiently transfected with WT and mutated (p.Phe508del and p.Val322Ala) pTCF. The maturated and non-maturated proteins were separated by gel electrophoresis. WT-CFTR gel separation, normally presents two bands (Fig. 1a). The first one is a band of approximately 140 kDa (band B), which represents the core-glycosylated protein located in ER, and the second is a band of approximately 170 kDa (band C), and represented mature, fully glycosylated protein that had migrated through the Golgi complex to the cell membrane.
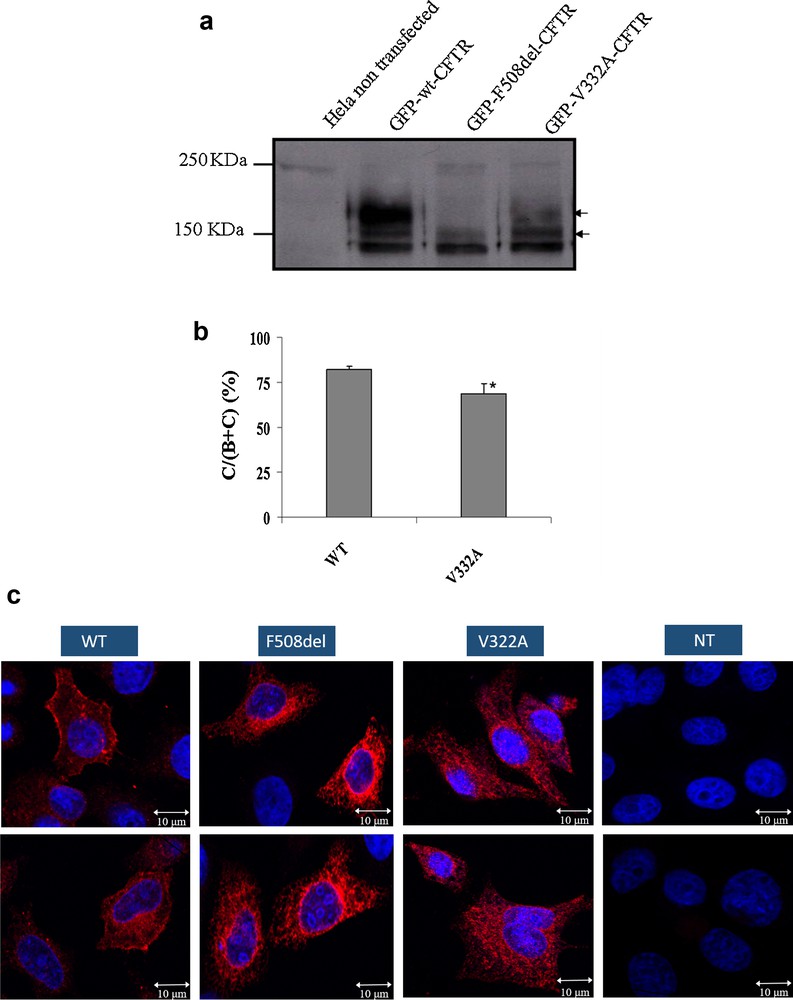
Impact of the V322A mutation on protein processing, localization and functionality: (a) Processing of WT and mutant CFTR proteins as assessed by the glycosylation status of the CFTR protein at the steady state on Western Blot. Hek293 cells were transiently transfected with WT, or mutants (p.Phe508del and p.Val322Ala). The p.Val322Ala proteins were obtained from three independent transfections. CFTR protein was detected by M3A7 (Chemicon), a mouse monoclonal antibody that recognizes an epitope at the C-terminal of CFTR in the region of residues 1370–1380. Arrows on the right indicate the positions of core-glycosylated (band B) and fully glycosylated (band C) forms of CFTR; (b) Quantitation of CFTR maturation efficiency calculated as the amount of mature CFTR (%C) relative to the total amount of CFTR produced (B + C). *, significantly different from the wild type (P < 0.05). (c) Subcellular localization of WT and mutant CFTR proteins in expressing HeLa and HEK 293 cells as assessed by confocal laser scanning microscopy. The green color results from the auto-fluorescence of the GFP and the red color results from the CFTR protein recognized by the Alexa Fluor 555® (Invitrogen). The numbers indicate the nucleus (1), endoplasmic reticulum (2) and cell membrane (3). The mutant p.Val322Ala (III) presents a membrane staining comparable to that of the WT (I)-transfected cells. A mean number of 15 cells were examined in three independent experiments for each CFTR protein analyzed. Scale bar, 10 μm.
For both WT and p.Val322Ala proteins, the bands B and C were observed, confirming a correct processing of the CFTR mutated protein (Fig. 1b). The band C is not observable for the p.Phe508del protein which is a class-II mutation and does not have an ER maturation.
3.2 The p.Val322Ala CFTR protein is correctly localized on the cell membrane
To evaluate the impact of the c.965T > C mutation (p.Val322Ala) upon protein localization, immunolocalization was used on HeLa and HEK 293 cells that had been transiently transfected with WT and mutated (p.Phe508del or p.Val322Ala) pTCF.
Cellular localization of proteins was visualized by confocal microscopy. Both WT and p.Val322Ala CFTR exhibited cell surface staining (Fig. 1c), whereas as expected the p.Phe508del CFTR that as retained in the endoplasmic reticulum (ER). These experiments indicate that the mutated p.Val322Ala protein is correctly localized at the cell membrane of these cells.
3.3 Normal functionality of the p.Val322Ala CFTR protein
We assessed the effect of Val322Ala substitution on the channel activity by iodide efflux assay. The mutated V322A CFTR transfected cells have the same activity stimulated by 10 μM forskolin and 30 μM Genistein like cells transfected by WT CFTR construct (Fig. 2). Furthermore, the cells transfected by K464 N have no CFTR activity as previously described before and as non-transfected cells [12].
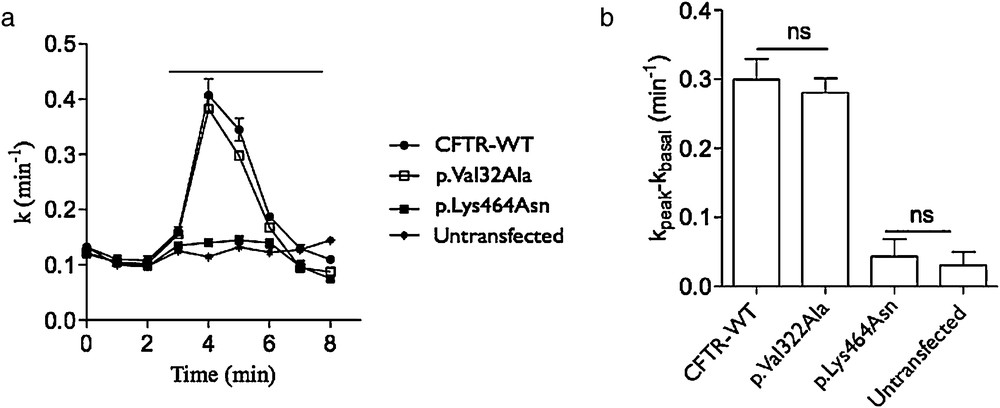
Impact of the p.Val322Ala CFTR mutation on protein activity. a) Time-dependent stimulation of iodide effluxes in cells transfected by the WT (CFTR-WT), c.1392G > T (p.Lys464Asn, class II as p.Phe508del) and c.965C > T (p.Val32Ala) pTCF plasmids. Channel activity was measured in the absence then in the presence of 10 μM forskolin and 30 μM genistein. The horizontal bar indicates the application of these drugs. b) Histogram showing the corresponding activity of each transfection. ns: non significant.
4 Discussion
The expected alterations of a CFTR mutation could vary from a total absence of the protein on the apical surface of epithelial cells to a reduction in the channel activity. The mutation might also have no notable consequences and should be considered as polymorphism. Therefore, understanding the possible effect of a CFTR mutation is essential to classify it in one or more of the six established classes or to consider it as a simple polymorphism.
In this paper, the c.965T > C mutation was studied to evaluate the potential alteration(s) of the mutated CFTR. The highly conserved Valine322 amino acid during evolution hypothesizes that its substitution may induce major consequences on protein processing and functionality. However, the substitution of this linear none polar hydrophobic amino acid by a residue that has the same characteristics, alanine, may reduce the impact of the mutation. Nevertheless, this valine322 is subject to another silent amino acid substitution by a methionine due to the c.964G > A mutation. The presence of this mutation in a CF patient highlights on the importance of the conserved valine at this position [15]. The in silico program indicates that the c.965T > C is “possibly damaging” by attributing a score of 0.88 over 1. Four other valine-to-alanine substitutions are already reported in different CFTR domains. The p.Val317Ala mutation is located in the fifth helix of the first TMD1 like the p.Val322Ala mutation. This mutation was identified through neonatal screening for hypertrypsinemia (Ferec et al., 1995), and the clinical symptoms vary from CBAVD with pancreatic sufficiency to pulmonary infection. Recently, this mutation was detected in a family that has one risk over four to have a child carrying this mutation in trans with the c.1521_1523delCTT (p.Phe508del) mutation. In addition, no protein studies have been conducted to determine the cellular consequences of this variant on CFTR. The other mutations are located in the intracellular loop linking the sixth helix of the TMD1 to the NBD1 (p.Val392Ala), in the NBD1 (p.Val456Ala) and NBD2 (p.Val1318Ala). Therefore, studying the impact(s) of the c.965T > C mutation using in cellulo analyses is essential for a prenatal genetic counselling.
The proper folding of the CFTR is essential to pass to the ER quality control system (ERQC) [16] and to engage the COPII vesicle machinery that ensures the transport to the membrane [17,18]. The co-translational formation of a partial CFTR (TMD1, NBD1, R-domain and TMD2) is sufficient to start the membrane translocation [9]. Interaction between the TMD1 and TMD2 is a major stage for obtaining the trafficable structure [19] and this is highlighted by a high number of diseases causing mutations found in the TMDs. Almost 300 mutations among the identified missense CF-causing mutation are localized in these domains [20]. The position of the Val322Ala substitution may disturb this TMDs crucial interaction. Therefore, the processing of the p.Val322Ala CFTR protein was evaluated by Western Blot analyses. A fully glycosylated band was observed as for the WT protein. The immunolocalization images obtained by confocal microscopy confirmed the correct processing of the mutated protein by showing cell membrane staining and not ER staining like for the F508del mutation. Therefore, the amino acid substitution seems to avoid conformational changes inducing a normal folding and processing of the mutated protein from the endoplasmic reticulum to the cell membrane.
However, essential residues ensuring a correct channel activity and ion selectivity are located in the MSDs [21]. In fact, while the helixes 1 and 6 of the MSD1 seem to have dominant role in CFTR folding and functionality, the helix 5 (containing Val322) may have a more minor role. However, this role remains more important than that of the helixes 2, 3 and 4, which perhaps play only a supporting role [22]. The mutation Val322Ala, in baby hamster kidney cells, showed a significant but light decrease of conductance [22]. Our channel activity test, in HeLa cells, revealed a non-significant decrease in ion transport across CFTR, indicating no influence of the Val322Ala amino acid substitution on the protein functionality.
5 Conclusion
In our test cell line, the obtained results showed that the Brittany c.965T > C variation may rather be a polymorphism than a mutation. However, regarding the important role of the conserved 322Val amino acid in the CFTR protein, preventive measures and clinical follow-up should be undertaken if the c.965T > C variation is associated in trans with another CF or CFTR-RD mutation.
Acknowledgments
The authors thank Anne Cantereau for her excellent assistance in confocal microscope. We also appreciate the valuable support of French Embassy in Cairo and Dr. Louis Moreau to AE. Raed Farhat received a fellowship from CNRS–Lebanon. This work was supported by ABCF2, Poitiers University Hospital and University of Poitiers, France.