

1 Introduction
Unambiguous taxonomic identification of biological objects is fundamental in any biological investigation. Inappropriate or uncertain description of a research object may not only lead to false results and inadequate conclusions, but also introduce serious misunderstandings in biological databases. These errors can be further replicated by the other researchers, causing even more discrepancies.
DNA barcoding is a standard, rapid and efficient method for identifying, discriminating, and discovering new species. This method may rely on a single or a combination of multiple DNA regions in the genome of the object under study [1,2]. Undoubtedly, this approach is convenient and very useful in identifying and classifying the world's biodiversity, in species delimitation, in food authenticity testing, in monitoring the illegal trade of wildlife or forensic investigations [3,4], as demonstrated in a number of previously conducted studies on plants [5,6] and animals [7,8].
Currently, much attention is paid to refining the DNA barcoding technique by searching for the most universal or most variable DNA barcode regions, discovering and characterizing new candidate regions, and determining the factors possibly affecting the final discrimination rate of the method. Successful application of DNA barcoding strongly depends on a selected region, the type of organism under study, phylogenetic distance and richness of the species in clades, but also on the size and completeness of the DNA barcode databases [9]. Moreover, in plant species, the discrimination rate depends on their lineages and equals 70% for angiosperms [10], and only 32% for gymnosperms [2]. DNA barcoding of gymnosperms is particularly challenging due to incomplete reproductive isolation among species, predominantly paternal inheritance of the chloroplast genome, hybridization and introgression, recent radiation and slow rate of molecular variation [11].
The vast majority of plant taxonomic investigations conducted with the DNA barcoding approach (including those mentioned above) were conducted on phylogenetically distant angiosperms, for which the species discrimination rate is relatively high. To date, little is known about the effectiveness of this method for discriminating closely related species (like conifers) and about the usefulness of DNA barcoding for investigations at the lower taxonomic level.
Usually, closely related conifers with similar morphology and lack of evident species determinants are grouped into larger units called complexes. The most well-known and intensively studied for years are the Pinus halepensis complex [12], the Pinus contorta-Pinus banksiana complex [13], the Pinus kesiya complex [14], and the Pinus mugo complex [15]. Research within such aggregates is particularly difficult, mainly due to complex taxonomic problems, with determination of origin, rank, presence of many synonymous names, similar ecological niches, as well as the occurrence of hybridization phenomena, combined with the presence of cross-species mixes. Species discrimination, delimitation or identification in such complexes is both scientifically interesting, but also practically very important with a view to the protection of endangered species, encompassed by such complexes.
The Pinus mugo complex comprises 16 species, 91 varieties, and 19 other forms [15]. This large and polymorphic complex of closely related pines natively occurs in the main European mountains, including the Pyrenees, the Alps, and the Carpathians [16]. Most of researchers agree that the Pinus mugo complex is comprised of three major components, i.e. Pinus mugo Turra (dwarf mountain pine), Pinus uncinata Rammond (moutain pine), and Pinus uliginosa Neumann (peat bog pine). Taxonomically, these three pines are considered to be either three independent species or subspecies inside Pinus mugo sensu lato [15–17].
Pinus mugo is a medium-sized shrub characterized by long and curved branches with symmetrical female cones [18,19], while P. uncinata is a tree reaching up to 12–20 m in height, with straight trunk and strongly asymmetrical female cones [20]. Pinus uliginosa is the most morphologically variable taxa out of the three pines under study, as it can occur as either a medium-size shrub (still usually much higher than P. mugo) or a mono- oligo- or polycormic (multi-trunk) tree with asymmetrical female cones [16,21].
In general, P. mugo is distributed in the eastern part of the Alps and in the Carpathian Mountains, while P. uncinata is commonly found in the western part of the Alps and in the Pyrenees. In turn, P. uliginosa forms several isolated and small populations in lowland peat bogs in Central Europe [16]. Moreover, in some unique stands, i.e. the Zieleniec reserve (the Sudety Mountains, southwestern Poland), several taxa from the Pinus mugo complex occur. Within such sympatric populations or contact zone due to the overlapping of phenological phases of the different pine taxa, the natural gene flow among them is observed as resulting in the generation of hybrid individuals [22–26].
The reciprocal genetic relationship among P. mugo, P. uncinata and P. uliginosa was extensively investigated using serological methods [27], allozymes [28,29] or, more recently, RAPD markers [20], molecular cytogenetics and flow cytometry [30] approaches. The obtained data showed a conserved genome organization and an absence of distinct genetic differentiation among them. Moreover, as recently demonstrated, these three pines share the same haplotypes of chloroplast and mitochondrial DNA [31,32]. Additionally, comparative study on needles and cones characteristics implied a lack of any powerful morphological marker for the straightforward identification of P. mugo, P. uncinata and P. uliginosa [33]. On the other hand, recent chemotaxonomic studies revealed some differences in composition of essential oils extracted from needles of P. uncinata and P. uliginosa [34]. Furthermore, based on mass spectrometry-assisted volatile compounds analysis, species-specific chemotaxonomic markers were proposed for these three pines [35].
Despite many investigations carried out and a number of various techniques used, no species-specific DNA markers have been developed for the three pines. Thus, the origin, species distinctiveness and relationship among P. mugo, P. uncinata and P. uliginosa, as well as their taxonomic status within Pinus mugo complex are still enigmatic and require further investigation to reach consensus. For these reasons, we decided to apply a DNA barcoding approach to get a further insight into this scientific problem. The current study covers:
- • sequencing eight chloroplast DNA barcode loci for P. mugo, P. uncinata and P. uliginosa individuals to enrich the barcodes database (deposited in GenBank);
- • evaluating a genetic variation level for these eight DNA barcodes among the three closely related pines in comparison to an external group of fourteen, more distant conifer species;
- • determining a species discrimination rate in Pinus mugo complex for a single and multiple DNA barcode regions;
- • gaining more insight into taxonomic relationships within the Pinus mugo complex;
- • assessing if any of the analyzed chloroplast DNA regions could be useful in resolving similar taxonomic ambiguities in the other pine complexes.
Consequently, we were able to verify the hypothesis about genetic distinctiveness of P. mugo, P. uncinata and P. uliginosa, and answer the question about the relevance of the DNA barcoding approach for discriminating closely related conifer species. The answer to the former question is particularly important – not only from a purely taxonomic point of view, but mainly in the context of the protection of those endangered taxa.
2 Material and methods
2.1 Taxon sampling
In this study, we analyzed seventeen different conifer taxa from the Pinaceae family, i.e. 14 species belonging to the Pinus genus and three species from the Picea genus (Table 1). The objects selection covered both phylogenetically distant and closely related taxa. In the case of 14 species, sequences of the eight chloroplast DNA barcode regions were directly downloaded from the GenBank database. The corresponding accession numbers are listed in Table 1. Due to lack of appropriate sequences for P. mugo, P. uncinata and P. uliginosa from the Pinus mugo complex, it was necessary to read these sequences in this study. To this end, the genetic material was extracted from pine needles of each one of these species, and appropriate regions were amplified and sequenced. Each species was represented by the three individuals available in the collection of the Dendrological Garden of the Poznań University of Life Sciences (Poland) with the following voucher codes: 07_0351_0001, 06_0351_0002 and 06_1204_0001 for P. mugo; 06_0790_0001, 06_0790_0004 and 06_0790_0005 for P. uncinata; 06_0526_0009, 06_0526_0008 and 06_0526_0007 for P. uliginosa.
A list of conifer species analyzed in this study.
| Genus | Subgenus | Section/subsection | Species | GenBank accession number |
| Pinus | Pinus | Sect. Pinus Subsect. Pinus |
||
| P. massoniana | NC_021439.1 | |||
| P. mugo | ||||
| P. sylvestris | JN854158.1 | |||
| P. thunbergii | D17510.1 | |||
| P. uliginosa | KX833097 | |||
| P. uncinata | ||||
| Sect. Trifoliae Subsect. Australes |
P. taeda | NC_021440.1 | ||
| Sect. Trifoliae Subsect. Contortae |
P. contorta | EU998740.4 | ||
| Strobus | Sect. Parraya Subsect. Cembroides |
P. monophylla | NC_011158.4 | |
| Subsect. Nelsonianae | P. nelsonii | NC_011159.4 | ||
| Sect. Quinquefoliae Subsect. Gerardianae |
P. gerardiana | NC_011154.4 | ||
| Subsect. Krempfianae | P. krempfii | NC_011155.4 | ||
| Subsect. Strobus |
P. lambertiana
P. koraiensis |
NC_011156.4 NC_004677.2 |
||
| Picea | P. abies | NC_021456.1 | ||
| P. morrisonicola | NC_016069.1 | |||
| P. sitchensis | NC_011152.3 |
2.2 DNA extraction, amplification and sequencing
Genomic DNA was isolated from needles of individual trees according to Doyle & Doyle's procedure [36]. The selection of the eight regions of chloroplast DNA, matK, rbcL, trnH-psbA, trnL-trnF, rpl20-rps18, trnV, ycf1, ycf2 was driven by their high genetic variability and informativeness for species delimitation, described in previously conducted studies [11,37–39]. Primers for amplification of rpl20-rps18, ycf2, trnH-psbA and matK regions were designed with PRIMER3 software [40], using complete chloroplast genome of Pinus uliginosa as a template [41] (Table 2). Primers sequences and reaction conditions used for amplification of rbcL, trnL-F, trnV and ycf1 were obtained from previous studies [37,42,43]. Polymerase chain reaction was carried out in a Veriti® Thermal Cycler (Applied Biosystems®, Life Technologies) using 100 ng of genomic DNA as a template in 20 μL of the reaction mixture, 1× PCR Buffer without MgCl2, 1 mM of each dNTP (Novazym, Poland), 0.5 mM of each primer (Sigma-Aldrich Co. LLC) and 1 U of HiFi Taq Polymerase (Novazym, Poland). The PCR products were resolved in 1.5% agarose gel in 1× TAE buffer, followed by staining with ethidium bromide, to verify the amplicons’ homogeneity and length. The PCR products were purified by ExoSAP-IT® (GE Healthcare, Cleveland, OH, USA) to remove non-incorporated primers, nucleotides, single-stranded PCR products, and residues of polymerase. The purified PCR products were sequenced in both directions with the same primers as those used for PCR using BigDye Terminator v. 3.1 CycleSequencing Kit (Applied Biosystems, Foster City, CA, USA) and analyzed with an ABI Prism 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer‘s instructions. All newly generated sequences were deposited in GenBank (Table 3).
Primers used for PCR amplification and sequencing.
| Region | Sequence 5’–3’ | Reference |
| rpl20-rps18 | F: ACTGCTAACCAACGGAAAGC | This study |
| R: CCTATTAGGTCGGGAGATCG | ||
| ycf2 | F: AAGTAATGATCCTGAGTGGAGTGAG | This study |
| R: TAAACGAAGACATAGATGGAAGGAG | ||
| trnH-psbA | F: ATTCACAATCCACTGCCTTG | This study |
| R: TAATGCTCACAACTTTCCTCT | ||
| matK | F: CCATAGATGCGGAAAGGAAG | This study |
| R: TCTGGGGAATATCTGAATGC | ||
| rbcL | F: GGACATACGCAATGCTTTAG | Wang et al., 1999 [37] |
| R: CCCTGCTTATTCCAAAACTT | ||
| trnL-trnF | F:CGAAATCGGTAGACGCTACG | Taberlet et al., 1991 [42] |
| R: ATTTGAACTGGTGACACGAG | ||
| trnV | F: GTAGAGCACCTCGTTTACAC | Wang et al., 1999 [37] |
| R: CTCGAACCGTAGACCTTCTC | ||
| ycf1 | F: GAAGGAAACAACAAATGTTCTAG | Parks et al., 2011 [43] |
| R: GATCCTCTCTGTTTATCGGGAA |
GenBank accession numbers of eight chloroplast DNA barcode regions for P. mugo, P. uncinata and P. uliginosa individuals.
| Taxa | Specimen voucher | matK | rbcL | trnH-psbA | trnL-trnF | rpl20-rps18 | trnV | ycf1 | ycf2 |
| P. mugo | 07_0351_0001 | MF193355 | MF193364 | MF193382 | MF193391 | MF193373 | MF193400 | MF193409 | MF193418 |
| 06_0351_0002 | MF193353 | MF193362 | MF193380 | MF193389 | MF193371 | MF193398 | MF193407 | MF193416 | |
| 06_1204_0001 | MF193354 | MF193363 | MF193381 | MF193390 | MF193372 | MF193399 | MF193408 | MF193417 | |
| P. uncinata | 06_0790_0001 | MF193359 | MF193368 | MF193386 | MF193395 | MF193377 | MF193404 | MF193413 | MF193422 |
| 06_0790_0004 | MF193360 | MF193369 | MF193387 | MF193396 | MF193378 | MF193405 | MF193414 | MF193423 | |
| 06_0790_0005 | MF193361 | MF193370 | MF193388 | MF193397 | MF193379 | MF193406 | MF193415 | MF193424 | |
| P. uliginosa | 06_0526_0009 | MF193358 | MF193367 | MF193385 | MF193394 | MF193376 | MF193403 | MF193412 | MF193421 |
| 06_0526_0008 | MF193357 | MF193366 | MF193384 | MF193393 | MF193375 | MF193402 | MF193411 | MF193420 | |
| 06_0526_0007 | MF193356 | MF193365 | MF193383 | MF193392 | MF193374 | MF193401 | MF193410 | MF193419 |
2.3 Sequence analysis and species discrimination rate
The obtained sequences were proof-read with CHROMAS Lite (version 2.01; Technelysium Pty Ltd). The edited sequences were aligned in MEGA 7 [44] program using ClustalX 2.0 tool [45]. The number of variable sites (VS), the number of parsimony informative sites (PIS) and genetic divergences (distances) for a particular chloroplast DNA barcode region and their combinations were calculated using MEGA 7 [44] according to the uncorrected p distance model at four taxonomic levels, i.e. genus (Picea and Pinus), subgenus (Pinus and Strobus), the closely related taxa (the Pinus mugo complex, PMC) and for all 17 conifer taxa under investigation. Additionally, the species discrimination rate in the closely related taxa from the Pinus mugo complex was evaluated both for single markers and the concatenated matrix using two different methods, i.e. the Tree-Building and the PWG-Distance methods. The Tree-Building method relies on the construction of Neighbor-Joining (NJ) trees for each single marker and the combination of barcode markers using the p distance model or the Kimura-2-parameter substitution model (K2P). According to Srivathsan & Meier [46], the p distance method is recommended for species-level discrimination in closely related groups. The bootstrap support for NJ trees was assessed using 1000 replicates. The sampled species was considered successfully discriminated if all individuals of a putative species formed a separate monophyletic clade (single cluster) in the phylogenetic tree with bootstrap support > 50% [2,10,47]. The ratio (in percentage, %) of successfully identified species inside the Pinus mugo complex in relation to all sampled species from this aggregate was calculated to assess the discrimination rate of the analyzed barcoding regions. The PWG-Distance method, recommended by the CBOL Plant Working Group [48], assumes that species discrimination is successful when the minimum uncorrected interspecific p distance in a species group is larger than the maximum intraspecific distance within each species.
2.4 Phylogenetic resolution
To assess the phylogenetic relationships within the 17 taxa under study, an NJ tree was constructed based on a concatenated matrix. The bootstrap support for NJ trees was assessed using 1000 replicates. The criteria for a species identification success were the same as in the Tree-Building method described above.
3 Results
3.1 Sequence characterization
In total, 184 sequences representing 17 conifer taxa belonging to Pinaceae were analyzed. Seventy-two new sequence reads for P. mugo, P. uncinata and P. uliginosa individuals were deposited in GenBank nucleotide sequence databases under accession numbers, as presented in Table 3.
The alignment lengths varied greatly among particular DNA barcode regions ranging from 507 bp for rbcL to 1296 bp for matK (Table 4) loci, while only small differences in their alignment length were observed at the genus and subgenus levels. An important difference in this parameter (of 150 bp) was observed only for ycf1 locus among Pinus genus and subgenus Pinus, excluding taxa from the Pinus mugo complex. On the other hand, significant differences in the alignment length were noted for a concatenated matrix composed of eight DNA barcode regions, which varied from 5300 bp for genus Picea to 5471 bp for genus Pinus (Table 4). The concatenated matrix analyzed in this study was 6001 bp in total length and represented approximately 5% of the Pinus uliginosa chloroplast genome with 119,877 bp (GenBank accession no. KX833097).
Genetic variation in eight chloroplast DNA barcode regions and concatenated matrix in conifer species, analyzed at four taxonomical levels.
| Region | Taxa group | AL (bp) | VS | PIS | p distance |
| matK | Genus Picea | 1296 | 7 | 0 | 0.004 |
| Genus Pinus | 1296 | 84 | 63 | 0.025 | |
| Subgen. Strobus | 1296 | 28 | 14 | 0.009 | |
| Subgen. Pinus (–) PMC | 1296 | 26 | 13 | 0.010 | |
| PMC | 1296 | 0 | 0 | 0.000 | |
| All 17 taxa | 1296 | 131 (10.11%) | 108 | 0.035 | |
| rbcL | Genus Picea | 507 | 15 | 0 | 0.020 |
| Genus Pinus | 507 | 24 | 12 | 0.013 | |
| Subgen. Strobus | 507 | 11 | 4 | 0.009 | |
| Subgen. Pinus (–) PMC | 507 | 11 | 4 | 0.010 | |
| PMC | 507 | 0 | 0 | 0.000 | |
| All 17 taxa | 507 | 33 (6.51%) | 22 | 0.017 | |
| trnH-psbA | Genus Picea | 566 | 11 | 0 | 0.013 |
| Genus Pinus | 605 | 67 | 33 | 0.032 | |
| Subgen. Strobus | 586 | 32 | 11 | 0.023 | |
| Subgen. Pinus (–) PMC | 599 | 20 | 1 | 0.014 | |
| PMC | 594 | 0 | 0 | 0.000 | |
| All 17 taxa | 615 | 114 (18.54%) | 80 | 0.055 | |
| trnL-trnF | Genus Picea | 594 | 6 | 0 | 0.007 |
| Genus Pinus | 628 | 38 | 27 | 0.025 | |
| Subgen. Strobus | 606 | 34 | 28 | 0.035 | |
| Subgen. Pinus (–) PMC | 622 | 20 | 7 | 0.016 | |
| PMC | 595 | 0 | 0 | 0.000 | |
| All 17 taxa | 628 | 77 (12.26%) | 58 | 0.039 | |
| rpl20-rps18 | Genus Picea | 513 | 6 | 0 | 0.008 |
| Genus Pinus | 511 | 29 | 22 | 0.026 | |
| Subgen. Strobus | 489 | 11 | 4 | 0.010 | |
| Subgen. Pinus (–) PMC | 511 | 3 | 2 | 0.003 | |
| PMC | 511 | 0 | 0 | 0.000 | |
| All 17 taxa | 515 | 57 (11.07%) | 48 | 0.039 | |
| trnV | Genus Picea | 530 | 5 | 0 | 0.006 |
| Genus Pinus | 528 | 14 | 10 | 0.011 | |
| Subgen. Strobus | 527 | 7 | 3 | 0.006 | |
| Subgen. Pinus (–) PMC | 528 | 5 | 3 | 0.005 | |
| PMC | 528 | 0 | 0 | 0.000 | |
| All 17 taxa | 530 | 25 (4.72%) | 20 | 0.016 | |
| ycf1 | Genus Picea | 831 | 7 | 0 | 0.006 |
| Genus Pinus | 876 | 283 | 229 | 0.157 | |
| Subgen. Strobus | 876 | 126 | 64 | 0.071 | |
| Subgen. Pinus (–) PMC | 726 | 32 | 11 | 0.021 | |
| PMC | 744 | 0 | 0 | 0.000 | |
| All 17 taxa | 882 | 347 (39.34%) | 281 | 0.164 | |
| ycf2 | Genus Picea | 936 | 134 | 0 | 0.102 |
| Genus Pinus | 1017 | 154 | 120 | 0.070 | |
| Subgen. Strobus | 954 | 59 | 33 | 0.031 | |
| Subgen. Pinus (–) PMC | 978 | 48 | 19 | 0.023 | |
| PMC | 972 | 0 | 0 | 0.000 | |
| All 17 taxa | 1028 | 235 (22.86%) | 197 | 0.089 | |
| Concatenated matrix | Genus Picea | 5300 | 185 | 0 | 0.024 |
| Genus Pinus | 5471 | 465 | 303 | 0.030 | |
| Subgen. Strobus | 5317 | 185 | 69 | 0.013 | |
| Subgen. Pinus (–) PMC | 5426 | 141 | 49 | 0.012 | |
| PMC | 5361 | 0 | 0 | 0.000 | |
| All 17 taxa | 6001 | 1017 (16.95%) | 810 | 0.059 |
According to the obtained results, individual DNA barcode regions differed significantly in the observed genetic variation. Amongst all 17 taxa under study, the highest number of variable sites (VS) and parsimony informative sites (PIS) was observed for ycf1 locus (347 and 281 sites, respectively). On the other hand, trnV turned out to be the least variable region with 25 VS and 20 PIS. The concatenated matrix contained 1017 of VS and 810 of PIS. The percentage of sequence variation varied from 4.72% for trnV to nearly 40% for ycf1, with 16.95% for the concatenated matrix (Table 4).
The sequence divergence, calculated according to the p distance model across individual and multiple DNA barcode regions at three different levels mentioned above is presented in Table 4. Considering all the regions combined, the p distance value calculated among all 17 conifer taxa under study reached 0.059, and varied greatly between particular regions ranging from 0.016 for trnV to 0.164 for ycf1. Sequence divergence in regions rbcL (0.017), matK (0.035), trnL-trnF (0.039), rpl20-rps18 (0.039), trnH-psbA (0.0055) was higher than in trnV (0.016), but significantly lower than in ycf2 (0.089) and ycf1 (0.164) regions.
The highest p distance value for individual regions at the genus level was observed for ycf2 in Picea (0.102) and for ycf1 in the Pinus (0.157) genera. At the subgenus level, the highest p distance value was observed for the ycf1 region in the subgenus Strobus (0.071) and for the ycf2 region in the subgenus Pinus (0.023). Apparently, in Pinaceae the ycf1 and ycf2 sequences evolved from 2 to 5 times faster than matK region, and from 4 to 10 times faster than trnV and rbcL regions. The sequence divergence was higher within two regions (rbcL and ycf2) in the genus Picea and within six regions (matK, trnH-psbA, trnL-trnF, rpl20-rps18, trnV, ycf1) in the genus Pinus. In conclusion, the subgenus Strobus is more variable than the subgenus Pinus, while the genus Pinus is more variable than genus Picea (Table 4).
On a closely related conifer taxa level, represented by P. mugo, P. uncinata and P. uliginosa from the Pinus mugo complex (PMC), no sequence variability was identified, either in individual or in multiple DNA barcode regions. Detailed comparison of individual DNA barcoding regions and combination of all chloroplast regions are shown in Table 4.
3.2 Species discrimination power and phylogenetic reconstruction
The discrimination power of the eight single DNA barcoding regions and of combination of all the regions among the three closely related pines from the Pinus mugo complex were determined using the Tree-Building and the PWG-Distance methods. According to the results obtained with the PWG-Distance method, the value of inter- and intraspecific p distance was zero both for individual and multiple DNA barcode markers (data not shown). Therefore, it was impossible to distinguish between P. mugo, P. uncinata, and P. uliginosa. Correspondingly, the results obtained using the Tree-Building method also showed a lack of taxa discrimination, since all the individuals representing these three taxa were grouped into one clade on the NJ tree with very high bootstrap support (100%) (Fig. 1).
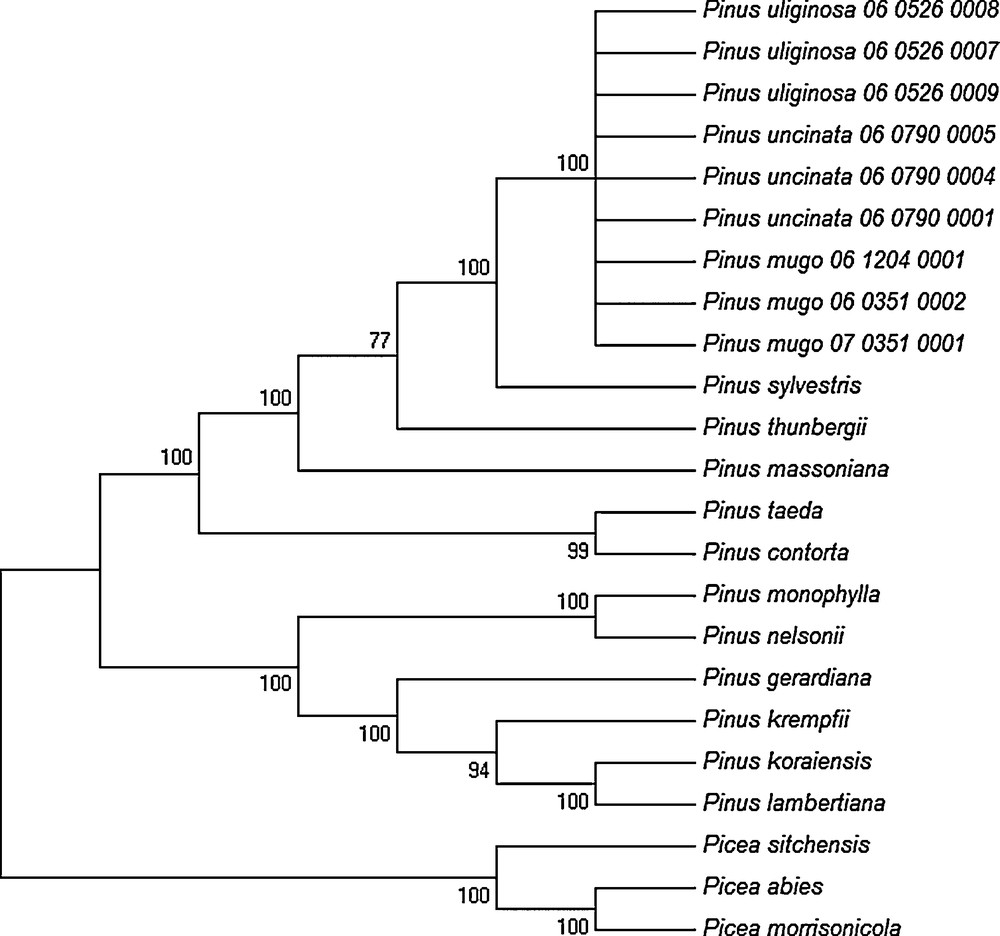
Phylogenetic (NJ) tree constructed for conifer species based on the concatenated matrix of chloroplast DNA barcodes analyzed in this study.
As shown in Fig. 1, all conifer species analyzed in this study, excluding the taxa from the Pinus mugo complex, were correctly discriminated and classified into the appropriate genus, subgenus and sections according to the commonly accepted taxonomy, with very high bootstrap scores (> 78% bootstrap support). The obtained NJ tree is clearly divided into two genera Picea and Pinus, with two distinct groups within the Pinus genus corresponding to two subgenera Pinus and Strobus. Moreover, the distinguished subgenera were clearly subdivided into individual sections and subsection. Importantly, all individuals representing the three closely related taxa from the Pinus mugo complex were localized in one large clade within the subsection Pinus.
The overall topology of phylogenetic NJ trees based on multiple eight chloroplast DNA barcode regions strongly supported the concept of monophyly of the genera Pinus and Picea (Fig. 1).
4 Discussion
In this study, we investigated the genetic divergence of eight chloroplast DNA barcode regions among 17 conifer taxa from the Pinaceae family, representing both phylogenetically distant and closely related taxa. We evaluated the effectiveness of the DNA barcoding approach by assessing the discrimination power of the individual and multiple DNA barcode regions for distinguishing the three closely related pines – P. mugo, P. uncinata and P. uliginosa belonging to the European mountain pines aggregate – to the Pinus mugo complex. To the best of our knowledge, this is the first study concerning the application of the DNA barcoding approach for resolving taxonomic and phylogenetic ambiguities in the highly polymorphic Pinus mugo complex.
In order to get a better insight into the genetic variation of the barcoding sequences, we analyzed the sequence divergence at four taxonomic levels, i.e. genus (Picea and Pinus), subgenus (Pinus and Strobus), closely related taxa (the Pinus mugo complex, PMC) and for all 17 conifer taxa together (Pinaceae). Eight selected chloroplast DNA barcoding regions represent traditional core barcodes as well as candidate sequences or supplementary regions. Traditional core barcodes, i.e. matK and rbcL, are universal and relatively frequently used, although characterized by limited genetic variability and relatively low discrimination power in the identification of closely related taxa [3]. In order to improve the discrimination power of the core barcodes, they are frequently used in pairs as two-locus barcodes (matK + rbcL). The candidate or supplementary regions, as for example trnH-psbA, ycf1 or ycf2, although characterized by significantly higher divergence level than the single and two-locus traditional core barcodes, are not that widely applied due to difficulties in designing universal primers for their amplification [3].
As demonstrated in the present study, the highest genetic divergence was observed for the ycf1 (0.164) region, when compared to the traditional core barcode markers, i.e. matK (0.035) and rbcL (0.017). Indeed, almost 40% of the ycf1 total sequence length was variable, which implies a high discrimination power for this particular region. The other supplementary regions ycf2 and trnH-psbA were characterized by slightly lower divergence (0.089 and 0.055, respectively). The obtained results clearly implied that the three candidate and supplementary regions under study demonstrate great potential towards conifer species discrimination and that ycf1 + ycf2 or their combinations (ycf1 or ycf2 with trnH-psbA) may serve as the new two-locus barcodes and efficiently complement the more universal but less divergent two-locus barcode region (matk + rbcL). A disproportionate amount of phylogenetic information and unusual evolutionary properties of the ycf1 and ycf2 regions were recently confirmed and highlighted by Parks et al. [38] based on analyses of 37 nearly-complete Pinus chloroplast genomes.
The comparison of the divergence level of the ycf1 and ycf2 barcoding regions within the individual Pinaceae genera revealed that while in the Pinus genus the former region is characterized by the highest divergence level, in the Piceae genus the most variable is the latter region. This may result from different evolution rates of these regions in the corresponding genera as well as different divergence periods between individual taxa. The precise determination of the reason for such high differences in the same regions in distinct genera of the Pinaceae family requires further, more detailed analyses. Nevertheless, the data presented here constitute a valuable suggestion for further studies for establishing the most efficient DNA barcoding region dedicated for the discrimination of species within the Pinus and Picea genera. Especially when previous studies on DNA barcodes dedicated for the Picea genus covering seven regions (matK, rbcL, rpoB, rpoC1, atpF-atpH, psbA-trnH and psbK-psbI) showed that none of them or their different combinations had sufficient resolution for spruce species discrimination, although matK + rbcL might be used as a two-locus barcode [11]. Interestingly, by comparison of the genetic divergence patterns generated by the DNA barcoding regions under study, we were able to systematize the barcodes according to their variability for the Picea and Pinus genera separately. For the genus Picea, the gradient from the most variable to the least variable was: ycf2 > rbcL > trnH-psbA > rpl20-rps18 > trnL-trnF > ycf1 = trnV > matK, while for genus Pinus, it was: ycf1 > ycf2 > trnH-psbA > rpl20-rps18 > matK = trnL-trnF > rbcL > trnV. Therefore, it might be concluded that for Pinaceae species identification, regions ycf1, ycf2, trnH-psbA and rbcL are more suitable, thus, they are more efficient than the rest of the regions under study. Correspondingly, the observed high variability of the ycf1 region has been also recently demonstrated in a study on the phylogenetic relationships and species delimitation within Pinus Section Trifoliae [49], as well as in reconstructing the infrageneric phylogeny of Pinus [38]. It altogether strongly suggests that indeed the candidate regions ycf1 and ycf2 may be particularly useful in the identification of species within Pinus and Picea genera, and serve as specific barcodes, dedicated to these taxa. According to a definition provided by Li et al. [3], a specific barcode is a fragment of DNA sequence that has a sufficiently high mutation rate to enable species identification within a given taxonomic group. In contrast, in a study by Armenise et al. [39], a 100% of total discrimination success in the identification of Italian conifers was achieved using the common rbcL + trnH-psbA, two-locus barcode, consisting of one core and one supplementary barcode region, which is not particularly specific to the present study.
The level of genetic divergence of matK and rbcL barcoding regions observed in this study (0.025 and 0.013) is comparable with the values previously determined by Wang et al. [37] based on the analysis of 32 pine species (0.022 and 0.012, respectively). The corresponding values were obtained for another two regions, i.e. trnV (0.011 vel. 0.011) and rpl20-rps18 (0.026 vel. 0.019) in the current study.
The number of variable (VS) and parsimony sites (PIS) for matK and rbcL determined in this study is highly corresponding to such results obtained by Syring et al. [50] during the analysis of twelve species belonging to Pinaceae. In the present study, 84 variable and 63 parsimony sites were identified in the matK region, and 24 and 12 sites in rbcL region, while Syring et al. [50] revealed 162 variable and 88 parsimony sites for the matK + rbcL combination. The observed differences in the VS and PIS values are relatively low and may result from the length of the analyzed sequences as well as from dissimilar species sets encompassed by the experiments. The moderate success of species discrimination (46.3%) with the core barcode markers (matK and rbcL) was achieved by Little et al. [51] during analysis of Podocarpaceae– the second largest conifer family. This indicates that the combination of matK and rbcL has lower discrimination power than the combination of matK or rbcL with the other regions like trnH-psbA, ycf1 or ycf2.
In general, the data obtained here as well as the previous literature reports mentioned above clearly demonstrate the high divergence level and the discrimination power of the eight DNA barcoding regions under study. However, only phylogenetically distant conifer taxa could be discriminated with these regions. With respect to the closely related taxa discrimination, application of the barcodes was unsuccessful. In spite of the high divergence amongst phylogenetically distant species, these regions were not useful in the identification of closely related taxa. Although we analyzed 6001 bp of the chloroplast genome, two times longer concatenated matrix than in standard phylogenetic studies conducted with typical bidirectional Sanger sequencing reads [37], we were still not able to discriminate the three closely related pines, i.e. P. mugo, P. uncinata and P. uliginosa from the Pinus mugo complex. Both inter- and intraspecific p distance values in individual as well as in multiple barcoding regions equaled zero. The absence of any genetic differences, i.e. substitutions, sequence length or nucleotide composition in the obtained DNA sequences among analyzed pine taxa from the Pinus mugo complex may result from recent diversification, introgression or/and low mutation rates of the plastid DNA barcodes inside this pine complex. Corresponding arguments were raised by Ran et al. [11] as potential reasons for difficulties in the discrimination of spruce species. Similar failure in the identification of species-specific genetic markers differentiating the taxa from Pinus mugo complex was also previously reported upon usage of RAPD [20] and SCAR markers [52].
The lack of identified and conserved genetic differences between P. mugo, P. uncinata, and P. uliginosa may suggest that they represent a single species with two or three taxa of subspecies level. To validate this, a large-scale comparison of more extensive chloroplast genomes, e.g., partial or complete plastome sequences, might be useful. The concept of exploitation of whole plastomes as the so called “super-barcodes” is gaining considerable attention [3]. This sophisticated approach was applied i.a. by Parks et al. [38] in the reconstruction phylogeny of Pinus based on the comparative analysis of 37 nearly-complete chloroplast genomes. Recently, the first complete chloroplast genome of a representative of the Pinus mugo complex – Pinus uliginosa – was published by Celiński et al. [41], which potentially opens the way to the application of the “super-barcode” strategy within this complex.
On the other hand, it cannot be excluded that P. mugo, P. uncinata and P. uliginosa may constitute separate species, and the nuclear genome rather than slowly evolving chloroplast or mitochondrial genomes should be scrutinized for identifying species-specific markers. Their recent divergence was supported by recent convincing data from a flavonic chemotaxonomy area. According to a “flavonic evolution” concept, taxa with the lower index of methoxylation (MeO), defined based on the relative contents of myricetin, isorhamnetin, larycitrin, syringetin, and prodelphinidin, represent more ancestral species than species with higher MeO index [53,54]. According to that study, the MeO index for P. mugo, P. uncinata and P. uliginosa equals approximately 34–37, while for the other species from this subsection, the MeO index is significantly lower, reaching 3 for Pinus massoniana and 4 for Pinus tabuliformis, through 13 for Pinus resiona and 17 for Pinus sylvestris, up to 32 for Pinus pinea. The species belonging to PMC were characterized by the highest values of the MeO index in the Pinus subsection. Interestingly, equally high MeO level (37) was observed for Pinus halepensis, classified into the Pinus halepensis complex [12]. Apparently, the high value of the MeO index is characteristic not only for new species, but similarities in its value may also constitute a specific determinant of closely related-species complexes.
Assuming recent divergence of P. mugo, P. uncinata and P. uliginosa driven by hybridization and introgression phenomena occurring in the PMC, identification of species-specific markers at the DNA level may appear highly challenging or even impossible, irrespective of the applied experimental strategy.
5 Conclusions
The eight-chloroplast DNA barcode regions analyzed in this study are useful for conifer taxa identification/discrimination of more phylogenetic distant species within Pinaceae family.
The analyzed markers are not suitable for distinguishing closely related pines from the Pinus mugo complex. In this specific case, the discrimination power of the barcoding regions under study equaled zero.
Apparently, extensive comparative studies applying whole plastome sequences and “super-barcode” strategy or even analyses of nuclear genome may support revealing current taxonomic ambiguities in complexes of closely related conifers, i.e. the Pinus halepensis complex, the Pinus contorta–Pinus banksiana complex, the Pinus kesiya complex or the Pinus mugo complex, and assist the development of specific barcodes dedicated to individual taxonomic groups.
Due to the lack of any species-specific differences at the DNA level at the concurrent observation of such differences at the secondary metabolite level, the problem of P. mugo, P. uncinata and P. uliginosa species distinctness, as well as their taxonomic status within the Pinus mugo complex, still remains unclear and requires further investigations.
Disclosure of interest
The authors declare that they have no competing interest.
Acknowledgements
This research was financially supported by the Polish Ministry of Science and Higher Education (No. NN304060339).