

1 Introduction
The worrisome selective increase in the incidence of hormone-dependent cancers in the last 25 years has led us to ask whether the increase in the number of EDs that have become ubiquitous in the environment is partially responsible.
This question confronts us with at least four difficulties.
- • There are many EDs. Over 350 synthetic molecules are present in the environment. They were developed because they have some advantageous characteristics (for instance, DDT for protection against malaria and the convenience of bisphenol A in food packaging). Many of these EDs can modulate the action or the metabolism of various hormones through different mechanisms.
Hence, each ED needs to be considered separately in terms of toxicity, mechanism of action and consequences of banning.
- • Carcinogenesis is a slow process, which can initiate very early (even in utero) under the action of initiator agents at the origin of a mutation or epigenetic modification, revealing itself late in life, often after the age of 50, under the effect of promotor agents, estrogens in the case of women and androgens in men. These agents will stimulate the growth of hormone-dependent cancers after puberty [1].
- • Because carcinogenesis is also a multifactorial process, it is difficult to assess from human epistemological studies the degree of responsibility of each of these factors.
- • Cancers exhibit considerable heterogeneity depending on the tissue affected.
2 Monitoring of cancer incidence trends in France since 1980
The analysis of cancer trends can help us identify the nature of the factors that are potentially responsible for cancers.
Since 1980, the incidence of cancers has differed according to cancer types[2,3]. Certain cancers that were frequent, such as gastric or cervix cancer, have decreased. Other cancers that are influenced by the action of various hormones have increased after 1980, in particular breast cancers until 2005 and prostate cancer until 2010, and some are still increasing as in the case of testicle and thyroid cancers. The increase in lung cancers among women is mainly due to increased tobacco addiction and the increase in melanoma is due to sun exposure. Lymphomas and melanomas are also on the rise. Pesticides are suspected to be the culprit. These cancers will not be discussed because none of them is considered to be hormone-dependent and not all pesticides have been proven to be endocrine disruptors.
This paper will be limited to hormone-dependent cancers and to the EDs that disrupt the activity of estrogens due to the fact that, like estrogens, their structures contain at least one aromatic ring.
The incidence of the most prevalent cancers, breast cancer in women and prostate cancer in men, increased the most between 1980 and 2005 [2]. Better screening is partly responsible for the rise in prostate cancer, but improved screening cannot explain the observed increase in breast cancer prior to the establishment of screening programs nor the increase in other cancers, such as ovarian and testicle cancers, which are not subject to screening. Furthermore, an increased incidence was observed for all age groups (including young individuals) (Fig. 1), indicating that the rise was not due to better screening or to aging of the population. According to French cancer records [3,4], the incidence of breast cancer started to decrease in 2005 and that of prostate cancer in 2010 (Fig. 2). These decreases are partially due to a diminished consumption of hormonal treatment for menopause in the case of breast cancer in France [5] and the USA [6], and to a significant reduction in screening based on plasma PSA levels in the case of prostate cancer.
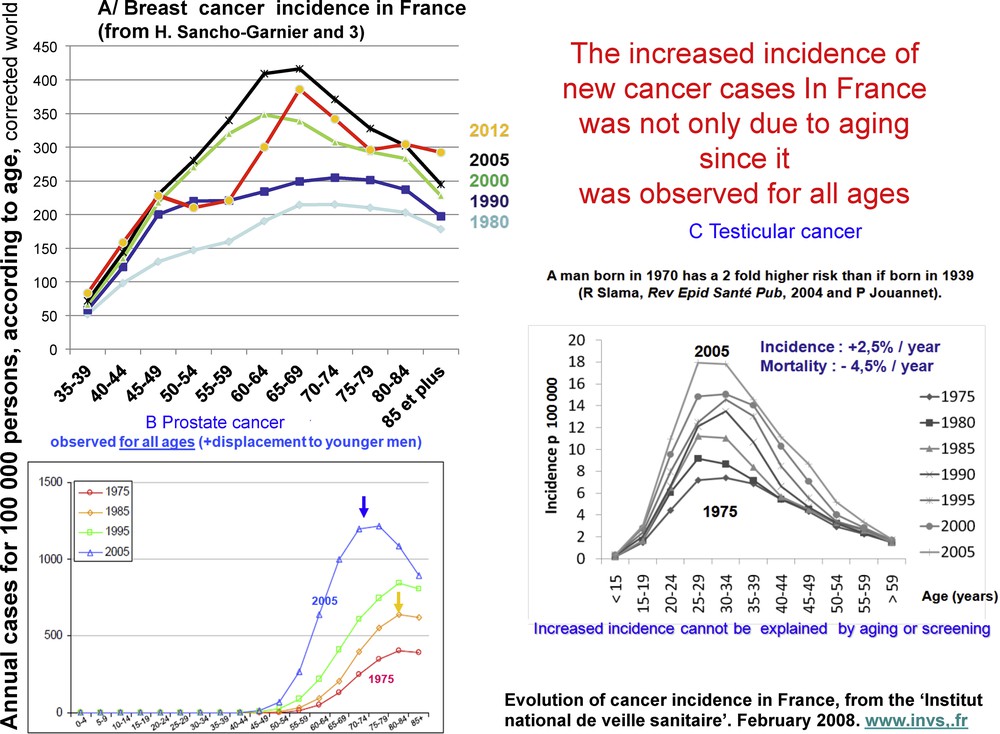
Age-independent increase in the incidence of three hormone-dependent cancers in France from 1980 to 2005. Evolution of epidemiological cancer data for 1980–2005, “Institut national de veille sanitaire”, février 2008. http://www.invs.fr.

Incidence and mortality of breast and prostate cancers in France from 1980 to 2012. After a clear period of increase, incidence decreased from 2005 for breast cancer and from 2010 for prostate cancer. This decrease followed a sharp decrease in the use of hormonal menopause treatment of breast cancer [5,6] and in the practice of PSA assays for prostate cancer.
Are EDs partly responsible for the high incidence of hormone-dependent cancers? What is their degree of responsibility for each cancer and what is the impact of each ED examined separately or in association?
I will mainly limit this discussion to breast cancer, which is still the leading cause of mortality from cancer among women in France [3], as well as in Europe [4]. It is, however, on track to follow the case of women in the USA, where it is now lung cancer.
The probable impact of the environment and/or food on breast cancer pathophysiology is demonstrated by the fact that the incidence of breast cancer increases in Asian women when they move to the USA.
3 A personal account of researches in the 1980s on the mechanism of action of estrogen in mammary carcinogenesis, showing how bisphenol A delayed this research
My colleagues at INSERM Unit 148 “Hormones and Cancers” and I had intense and sometimes heated debates with our competitors, who were mainly American, on the effect of estrogens on mammary carcinogenesis.
At the time, it was unknown whether estradiol promoted proliferation by acting directly on mammary cells or indirectly via other cells or tissues (Fig. 3). This was due to the fact that the in vivo effect of estradiol (E2) on the growth of mammary tumors in rodents was readily reproducible, while researchers had difficulties observing a reproducible in vitro mitogenic effect on mammary cancer cell line grown in plastic bottles.
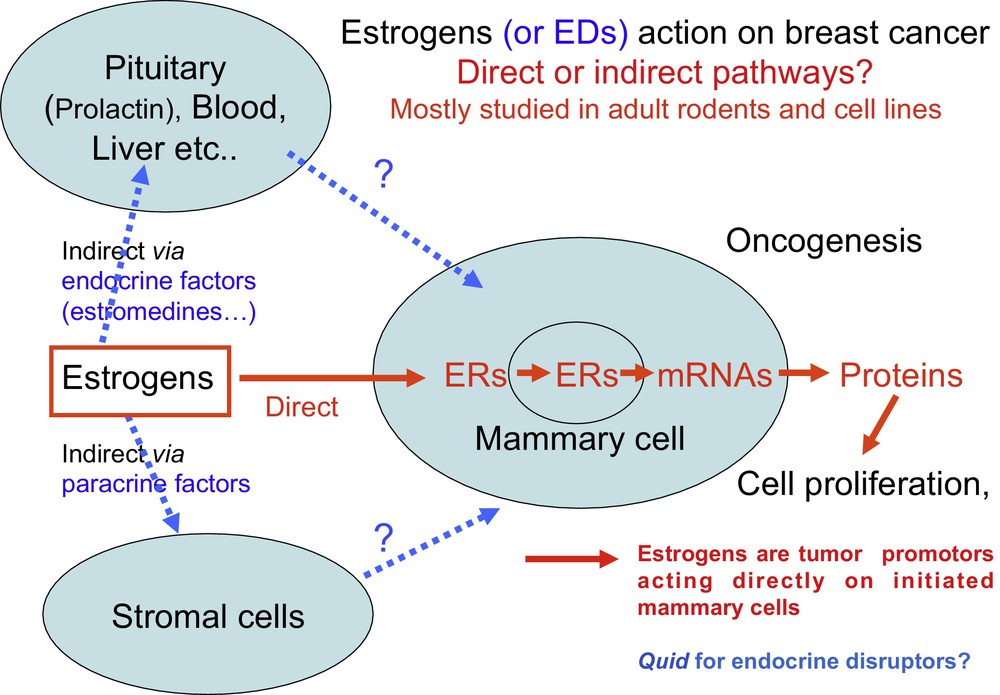
The two main hypotheses for direct and indirect action of estrogens formulated in the 1970s and 1980s are still current regarding the in vivo effect of many EDs. Since 1990, it has been generally acknowledged and demonstrated that estrogens have a direct effect on mammary cells that have already initiated a process of carcinogenesis [1]. For most EDs, including bisphenol A, whether they act directly or indirectly and at what developmental stage remains unknown.
Hence, two major hypotheses were formulated: estrogens act directly on mammary cells or they act indirectly via other cells or plasma factors. Several American groups favored an indirect effect through prolactin or pituitary estromedin [7] or other plasma factors named estrocolyones [8]. In the 1980s, we [9] and Marc Lipmann's laboratory [10] favored a direct effect because we observed a mitogenic effect on two breast cancer cell lines (MCF7 et T47D) that expressed the estrogen receptor α (ER + ). However, the cell culture media should be devoid of any compounds with estrogen activity either in the serum or in the growth media.
The fact that the mitogenic effect was not reproducible was due to the presence of estrogens in the growth medium of control cell cultures (no estradiol supplement). The possible culprits were the phenol red dye (a pH indicator), which could be easily eliminated and some factor released by certain batches of plastic bottles. The latter was subsequently identified as being bisphenol A by Krishnan Av et al. [11]. Furthermore, we observed that the estradiol mitogenic effect in T47D cells was at its peak at 1 nM and decreased from 10 nM concentrations. The mechanism underlying this biphasic effect, also observed for some of the EDs, has not been identified [9].
The direct mitogenic effect of estrogens on mammary cells via hormone receptors responsible for their tumor promoting activity was then endorsed. But the direct action of most EDs has yet to be demonstrated in vivo.
4 Three ED examples among those most studied as carcinogenic in women: distilbene, bisphenol A and DDT
These three molecules bind to estrogen receptors (ER), albeit with quite different affinities. Most EDs (except distilbene) are weak estrogens and anti-androgens that bind ERs with a very weak affinity at micromolar concentrations. DDT (dichlorodiphenyltrichloroethane) and other persistent organochlorides, such as PCBs (polychlorinated biphenyls) and dioxins, are hydrophobic and accumulate in the environment, fats and the food chain (half-life of ∼10 years). They have been shown to have an estrogenic effect on aquatic animals. By contrast, bisphenol A is water soluble and rapidly metabolized, but it is ubiquitous in the environment, food and living organisms.
4.1 DDT, an organochlorine insecticide
DDT, an organochlorine insecticide, was extensively used from 1940 to 1970 against mosquitoes to fight the diseases that they transmit to humans, such as malaria.
Briefly, arguments concerning its toxicity have evolved in three steps:
- • from 1945 to 1970, caution was recommended for humans as DDT persistence in the environment was proven responsible for the estrogenic effects observed in aquatic animals (sex change) and sterility in birds (see R. Carlson's book “Silent Spring”, published in 1962). Hence it was banned in the USA in 1970;
- • from 1970 to 2010, none of the epidemiological studies in adults (studies of cases/control, meta-analyses, etc.) could confirm a carcinogenic effect in humans [12,13];
- • it was only in 2011 that the results from large California cohorts, which were followed for more than 50 years and included data of plasma DDT levels at the time of exposure, showed that there was a probable risk associated with prenatal or perinatal exposure to DDT. Serum concentrations of DDT and its DDE metabolite, tested in various European and American cities after the ban on DDT, showed a rapid decrease in DDT while DDE diminished very slowly [14]. In the first cohort of 20,500 children and young women exposed to DDT between 1945 and 1960 and followed over more than 60 years, Barbara A. Cohn showed in 2011 [14] that the risk of breast cancer varied both as a function of the age of the child in 1945 at first exposure and of DDT serum concentrations measured in 1960. The risk was five times higher for high DDT concentrations, but only for the exposed girls under 10 [14]. Exposure after puberty did not increase the risk. In the second cohort of 20,700 pregnant women [15], the environmental exposure of the mothers to DDT from 1959 to 1967 increased, fifty years later, the risk of breast cancer in 9,300 of the daughters by a factor of five compared to the non-exposed mothers, but this effect was observed only in the case of high DDT concentrations. Breast cancers were ER+ (83%), PR+ (76%) and HER2− (59%), but there were four times more HER2+ cancers when DDT levels in mothers were high. Two other studies confirmed that DDT toxicity depended on the precocity of exposure [16,17]. Interestingly, no low-dose effect was observed with DDT in contrast to other EDs such as bisphenol A.
The study of the Seveso cohort came to the same conclusion: only young women had a higher risk of breast cancer following the accidental exposure to dioxin while dioxin anti-estrogen activity might protect adult women [18].
4.2 Lessons from distilbene: from women to mice and inversely
Distilbene (diethylstilbestrol, DES), a synthetic compound with a strong affinity for ERα, was the first proven carcinogenic ED in humans.
In utero exposure to DES, given to mothers to treat hormonal sterility due to estrogen deficiency, induced gynecological cancers in daughters and granddaughters (F1 and F2). The increased risk of breast and endometrial cancer was hard to establish due to the high frequency of these cancers. It was the occurrence of a clear cell adenocarcinoma of the vagina, a very rare cancer that led to the demonstration of a causal link between cancer and distilbene treatment of mothers. As early as 1971 in the USA, this led to banning the use of distilbene during pregnancy [19].
Mice studies confirmed this causal link and made it possible to study its mechanism. The first molecular target was identified. The double inactivation of the ERα gene suppresses the carcinogenic effect of distilbene [20]. Inversely, from this observation, one can consider that in vivo rodent models are useful to understand carcinogenesis in humans. It was then shown that distilbene interferes with the normal development of estrogen target tissues through genetic and/or mainly epigenetic (by CpG methylation for instance) mechanisms. Altered development is an early stage in carcinogenesis that only reveals itself following puberty under the promotor effect of ovarian estrogens.
4.3 Bisphenol A
The effects of bisphenol A on breast and prostate cancers were mainly studied by North American and Danish laboratories, see the various reviews in [21,22] as well as the consensus of the American Endocrine Society [23], which was confirmed in 2012 [24].
The bisphenol A structure contains 2 phenol groups, like distilbene, but in a different spatial orientation, which explains its extremely low affinity for ERα and its low estrogenic activity. Its general ban, proposed in France in spite of the more nuanced opinion of the French Academy of Medicine [25], is still a source of debate in Europe because substitutes, except for glass baby bottles, are not yet available. Each year, several tons of bisphenol A are synthesized worldwide. Bisphenol A is ubiquitous in food and the environment, which makes it hard to define a control population in epidemiological studies.
- • Its polymer has been used for 50 years in food packaging (polycarbonates and epoxy resins in food cans) and in some dental cements and cash register receipts. Bisphenol A released at high temperatures or pH extremes is potentially toxic. Adults rapidly eliminate it as a glucuronide conjugate after inactivation by the liver, but it is eliminated much more slowly by fetuses and young children.
- • Continuous human exposure (as monitored in plasma, urine, milk and amniotic liquid) is usually much higher in young children. Its level varies with nutrition and has been found to be close to the ones observed in rodents and monkeys that develop pre-carcinogenic mammary lesions in experimental studies. The mean bisphenol A concentration in 1764 French pregnant women who were part of the Elfe cohort and gave birth in 2011 was 870 μg/kg. This level exceeds the threshold authorized by government agencies (French Agency for Food, Environmental and Occupational Health Safety (ANSES), Santé publique France).
- • A pre- and perinatal mitogenic effect of bisphenol A on rodent mammary glands was found in all studies of mice and rats, whatever the method of administration. It resulted in a significant increase in pre-carcinogenic mammary lesions (ductal hyperplasia) and in situ ductal cancers provided that xenoestrogens such as bisphenol A released by the cages or present in the feed were excluded [26–29].
- • Some studies showed that bisphenol A was a more powerful mammary co-carcinogen at 0.25 μg/kg than at higher doses [25]. This low-dose effect (< 0.25 μg/kg) was also observed after oral administration in transgenic MMTV-HER2/Neu mice [27]. This low-dose effect has been observed for other EDs but not DDT (see above). This makes the definition of a daily authorized exposure for humans by public agencies more complicated. For bisphenol A, the dose was recently decreased to 5 μg/kg/day in Europe, as in the USA and Canada, which is inconsistent with its low-dose effect.
- • While the toxicity of early (pre- or perinatal) exposure to bisphenol A, especially in rodents, is real, its mechanism remains unknown in spite of many studies. Different targets (receptors and enzymes) could be involved at different doses, as well as a negative feedback regulation of receptors at higher doses. However, its target is likely to be different from that of distilbene because (1) bisphenol A affinity for nuclear hormone receptors is 1000 times lower; (2) ERα KO mice, which become insensitive to the carcinogenic effect of distilbene, remain sensitive to the deleterious effect of bisphenol A on Leydig cells [30].
- • Bisphenol A could act through other nuclear or membrane (possibly GPR30) receptors, or even other proteins (such as CYP 450 enzymes) and modify the sensitivity to estrogens during development through an early mechanism, which could be epigenetic [31].
Because the cellular and molecular targets are unknown, the development of rapid and reproducible assays, in particular in vitro tests to screen for substitute products that are as effective but not toxic, are hampered. This should be a priority for researchers and the food industry.
5 Conclusions
Converging experimental approaches, both in vitro experiments on cell lines and in vivo on animals, combined with epidemiological studies monitoring cohorts of women exposed to EDs make a compelling case for their co-carcinogenic effect, an effect that does not become evident until much later in life. For example, the effect of bisphenol A and DDT on breast cancer after early in utero or perinatal exposure appears much later after puberty (Fig. 4). Other EDs (including PCBs, parabens, dioxins and certain pesticides) are co-carcinogens with a delayed effect on breast and prostate cancer [32], but they probably may have a different mechanism of action.
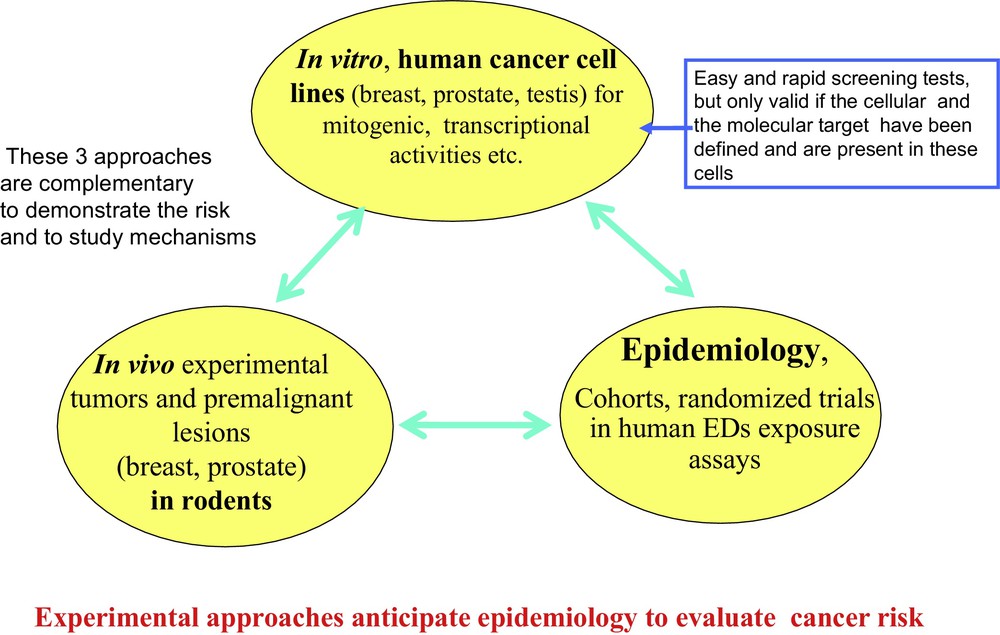
The three approaches used to study the action mechanism of an ED on carcinogenesis. Results from epidemiological studies should agree with experimental (in vitro and in vivo) studies. However, epidemiology is more complex and results are much slower to obtain. Very often, large cohorts of parents and their descendants need to be monitored over decades, in conjunction with assaying EDs (and/or their metabolites) during the initial ED exposure of parents.
Specifying the extent to which each ED is responsible for carcinogenesis cannot be done because of the high level of potential toxic compounds, their variability in time and their persistence in the environment and the food chain. This is made more difficult by the fact that there are other factors that may contribute to the increased incidences of these cancers (diet, lifestyle, better screening, increasingly postponed pregnancies in the case of breast cancer, among others) [33].
The toxicity of each molecule was probably low when assessed in isolation and in adults, but might be higher when assessed in combination (see the cocktail effect in reference [34]) and during early exposure (infants, pregnant and nursing women, for whom protection should be a priority). The degree of toxicity also varies depending on the genetic background of each individual, which was shown in several studies to modify sensitivity to hormone and ED mixtures.
A better understanding of the in vivo mechanism of action of each ED during development would help researchers, in conjunction with industry, to develop substitute products that are just as efficient and less toxic.
The toxicity of a molecule as evaluated by epidemiologists and toxicologists should guide legislators and not its classification as an endocrine disruptor, which is based on its mechanism of action rather than its risk. Toxic effects have not been always proven to be due to hormonal disruption. For instance, some pesticides such as glyphosate are toxic, although hormonal disruption is not proven [35]. Some EDs can have beneficial effects. Depending on their concentration, genistein, a soy constituent eaten by Asian people, and other phytoestrogens can have a protective effect on hormone-dependent cancers [36], but be toxic for male reproduction. Hence, EDs are not all potential carcinogens, some are innocuous or protective at low doses as in the case of soy phytoestrogens. Legislators should refrain from legislating on the basis of in vitro ED classification tests when the in vivo toxicity mechanism is unknown. Each molecule should be considered separately in terms of mechanism and toxicity. Moreover demonstrating in vivo toxicity in human will require the implementation of lengthy and expensive tests.
Disclosure of interest
The author declares that he has no competing interest.
Acknowledgements
The author is grateful to Prof. Hélène Sancho Garnier for the French epidemiological data, to the members of the “Groupe de travail bi-académique médecine/pharmacie sur les pertyurbateurs endocriniens” (2011) as well as to Jean-Yves Cance U1194, INSERM, for providing the figures.