


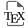
 CC-BY 4.0
CC-BY 4.0
1. Introduction
Defects in the complex process of cardiac morphogenesis underlie congenital heart defects (CHD) that affect 1 in 100 live births [1]. The heart is derived from mesoderm, the central of the three embryonic germ layers, and originates in the anterior region of the early embryo as a tubular structure with venous (inflow) and arterial (outflow) poles. Cardiac progenitor cells located in splanchnic mesoderm, overlying future foregut endoderm, contribute progressively to growth of the heart. These cells, known as the second heart field (SHF), ultimately give rise to ventricular septal, right ventricular and outflow tract myocardium at the arterial pole, and atrial, including atrial septal myocardium, at the venous pole. Deployment of SHF cells is coincident with looping morphogenesis, an essential conserved process during which the heart tube rotates to the right to correctly position the future regions of the definitive heart. Subsequent steps of heart development involve cardiomyocyte proliferation driving chamber morphogenesis, development of the coronary arteries and cardiac conduction system coordinating the heartbeat, and cardiac septation. During septation the formation of ventricular and atrial septa generates the four chambered heart, and the embryonic outflow tract is divided into the outlets of the left and right ventricles, the ascending aorta and pulmonary trunk. The initially tubular embryonic heart thus acquires independent right atrioventricular and left ventriculoarterial junctions. Cardiac septation is complete at birth when the pulmonary duct and the interatrial foramen close, coincident with pulmonary perfusion, isolating the pulmonary and systemic circulatory systems.
Heart tube elongation by addition of SHF cells creates the template for cardiac septation and therefore perturbation of SHF deployment in human patients and animal models results in a spectrum of common forms of CHD [2, 3]. These include ventricular and atrial septal defects as well as outflow or conotruncal defects, and lead to failure to correctly separate the systemic and pulmonary circulation. Dissecting the developmental programs driving normal cardiac progenitor cell deployment is thus essential to understand how defects in these processes result in CHD, as well as for directing stem cell derived cardiomyocytes to different regional fates for drug testing and tissue repair following cardiac damage. Here we review the contribution of the SHF during heart development and its involvement in CHD, with focus on the roles of two T-box transcription factors implicated in human CHD, TBX1 and TBX5.
2. Early heart development and the second heart field
Nascent mesodermal cardiac progenitor cells emerge from the primitive streak at gastrulation and migrate to the anterior lateral region of the embryo (Figure 1A). Two distinct waves of progenitor cells have been identified that independently activate the gene encoding the early mesodermal transcription factor MESP1 [4, 5, 6]. The first wave generates first heart field (FHF) progenitor cells that differentiate to form bilateral cardiac primordia that converge in the ventral midline as the foregut closes, giving rise to the cardiac crescent and ultimately to left ventricular and atrial myocardium.
Cardiac development. Cartoon showing the contribution of first (pink) and second (green) heart field progenitor cells to the developing mouse heart. (A) Heart tube morphogenesis, (B) heart tube looping and elongation and (C) chamber growth and cardiac septation. FHF, first heart field; SHF, second heart field; L, lateral; M, medial; A, anterior; P, posterior; D, dorsal; V, ventral; AP, arterial pole; VP, venous pole; HT, heart tube; RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; NT, neural tube; DM, dorsal mesocardium; OFT, outflow tract; Ao, aorta; PT, pulmonary trunk; E: mouse embryonic day, D: human embryonic day.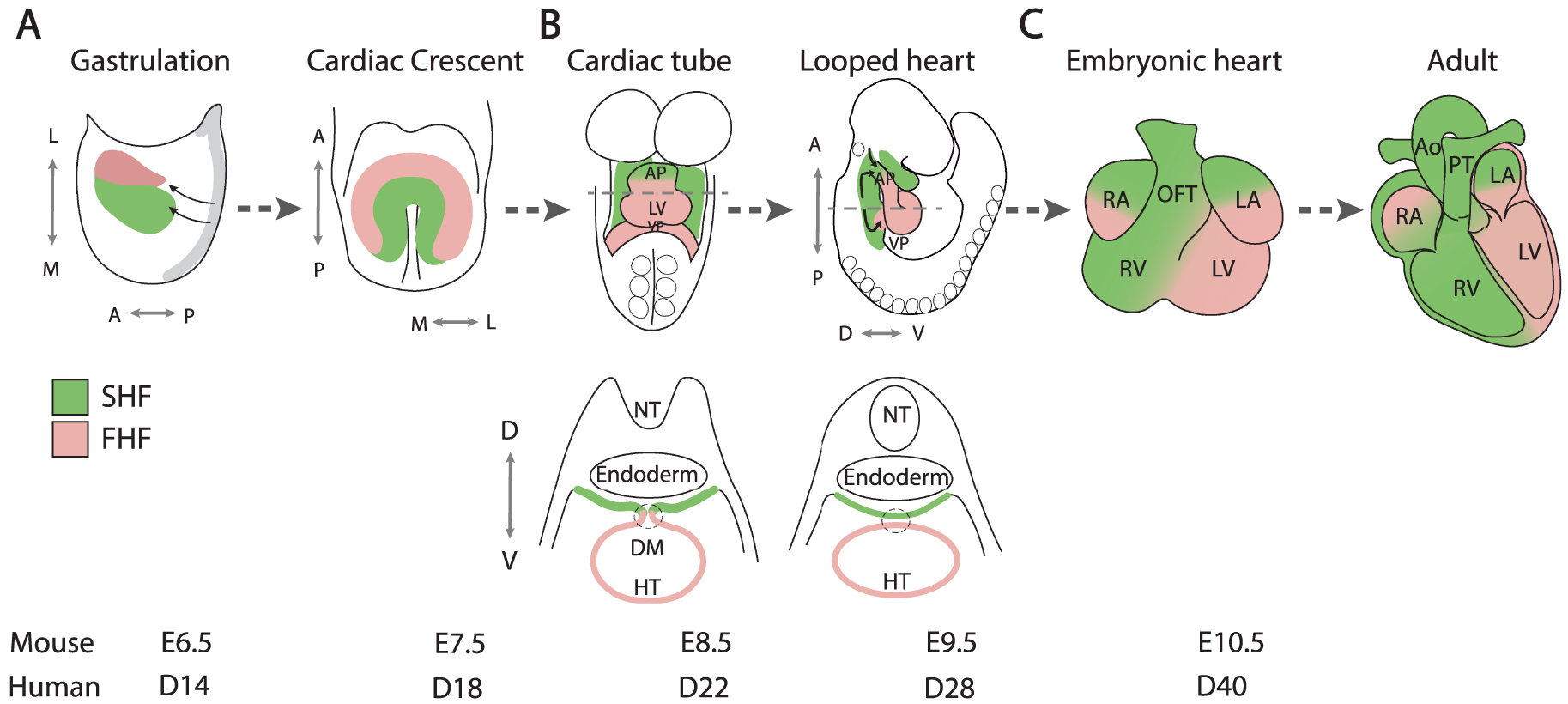
The cardiac crescent is transformed into a transiently linear heart tube that subsequently elongates and loops to the right, generating the embryonic heart at midgestation in the mouse, equivalent to the 5th week of human development (Figure 1B). Heart tube elongation is driven by the progressive addition of SHF cells, derived from the second wave of Mesp1 expression, located in pharyngeal mesoderm dorsomedial to the early heart tube, including the dorsal wall of the pericardial cavity [5]. The SHF was first identified in 2001 through analysis of a Fgf10 enhancer trap transgene expressed in murine pharyngeal mesoderm [7] and by cell labelling experiments in avian embryos [8, 9]. These findings built on pioneering embryological evidence for the addition of extracardiac cells to the growing cardiac poles [10, 11]. Initially contributing along the entire length of the cardiac primordium, SHF addition becomes restricted to the arterial and venous poles after breakdown of the dorsal mesocardium separates the heart tube from the dorsal pericardial wall (Figure 1B). Genetic tracing and retrospective lineage experiments have shown that the first and second heart fields correspond to distinct genetic lineages defined by their exclusive contributions to the outflow tract and left ventricle respectively, and that SHF progenitor cells adding to the arterial and venous poles are clonally related [12, 13, 14]. Subsequent labeling experiments and genetic lineage analyses have revealed the progressive addition of SHF progenitor cells to the elongating heart tube, giving rise successively to the right ventricle, proximal and distal outflow tract, and atrial myocardium, as well as smooth muscle cells of the pulmonary trunk (Figure 1C) [15, 16, 17, 18]. Clonal relationships have been established between these SHF subpopulations and branchiomeric skeletal muscles of the head and neck, consistent with the existence of common cardiac and skeletal muscle progenitor cells in pharyngeal mesoderm [19, 20, 21, 22]. This is supported by the recent identification of multilineage primed progenitor cells that contribute to skeletal muscle and myocardium, using single-cell RNA-seq analysis [23]. Single-cell RNA sequencing is a revolutionary approach that has yielded unprecedented insights into heterogeneity and developmental trajectories of cardiac progenitor cell populations [24]. These include the identification of a population of FHF cells termed the juxtacardiac field, located at the lateral boundary between the embryo and extraembryonic mesoderm, that contributes to growth of the early cardiac primordium along its ventrolateral margin, in contrast to SHF contributions along the dorsomedial margin [25, 26, 27]. Single-cell transcriptomics has also revealed differences in gene expression between left and right SHF cells, consistent with the recent finding that the SHF is a driver of looping morphogenesis [28, 29]. Defects in cardiac looping, or in the prior establishment of embryonic laterality, have been implicated in a spectrum of CHD [29, 30]. Dynamic imaging using two-photon and laser scanning fluorescent microscopy have begun to confirm the trajectories taken by cardiac progenitor cells during growth of the early heart. These experiments have revealed a temporal delay between first and second heart field cell deployment and distinct patterns of cell behavior in the juxtacardiac and second heart fields during heart tube assembly [6, 31]. In addition, quantitative morphometric approaches provide high-resolution understanding of the complex events associated with early heart development [32].
3. TBX1 and arterial pole development
Progenitor cells adding to the arterial pole of the heart, known as the anterior SHF, give rise to right ventricular and outflow tract myocardium. As these cells converge on the growing arterial pole, they move through a transition zone in which progenitor cell gene expression is downregulated and myocardial differentiation activated (Figure 2A) [33, 34]. While lack of SHF deployment results in severe early lethal phenotypes, milder defects in this process generate an outflow tract that is too short to allow the normal alignment between the left ventricle and ascending aorta during cardiac septation. Consequently, anterior SHF defects contribute to conotruncal CHD, that account for 30% of CHD, ranging from partial ventriculoarterial misalignment as in the case of overriding aorta and tetralogy of Fallot, to double outlet right ventricle (Figure 2B). Defective growth of the ascending aorta has also been implicated in transposition of the great arteries [35].
Arterial pole development and outflow tract septation. (A) While SHF cells contribute to the myocardial wall of the outflow tract at the transition zone, SHF addition is regulated by cardiac neural crest cells that also mediate outflow tract septation. Embryonic laterality impacts on rightward looping and ventriculoarterial alignment. (B) An example of a conotruncal congenital heart defect, tetralogy of Fallot. SHF, Second heart field; AP, arterial pole; VP, venous pole; A, Anterior; P, Posterior; D, Dorsal; V, ventral; RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; OFT, outflow tract; Ao, aorta; PT, pulmonary trunk.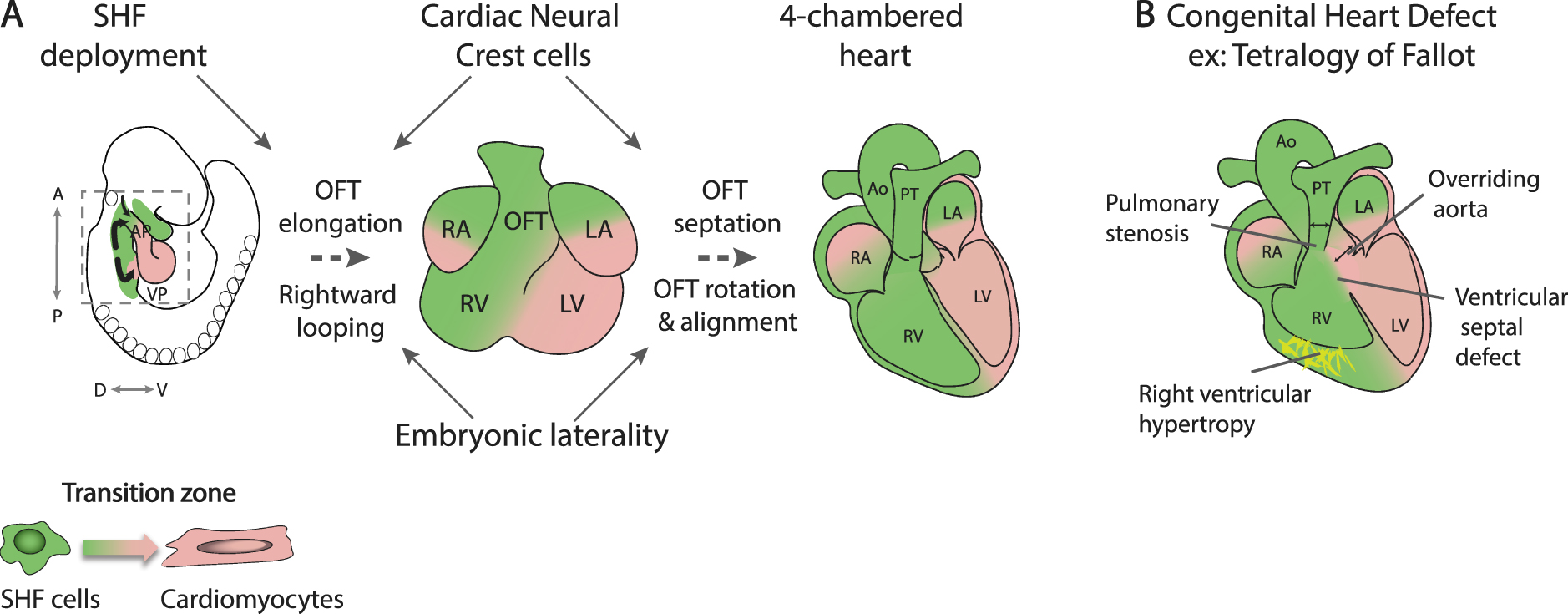
Work in many groups worldwide has identified critical transcriptional regulators, intercellular signaling pathways and epithelial features that maintain SHF progenitor cell status and regulate progressive differentiation at the cardiac poles [36, 37, 38, 39]. In addition to Fgf10 expression, SHF cells are demarcated by expression of the transcription factors ISL1, TBX1 and an enhancer from the Mef2c locus [7, 15, 16, 40]. These transcription factors integrate the activity of signaling events including autocrine signaling from pharyngeal mesoderm as well as signal exchange with surrounding cell types, in particular adjacent ventral pharyngeal endoderm and neural crest-derived mesenchyme. Heart tube elongation is regulated by a balance between pro-proliferative FGF signals and pro-differentiation BMP signaling; FGF10 expression in the SHF, for example, acts together with FGF8 to control arterial pole development [41]. As SHF cells differentiate in the transition zone, Fgf10 expression is downregulated by NKX2-5, that blocks ISL1-mediated activation of an intronic Fgf10 enhancer [42]. SHF cells in the dorsal pericardial wall have been shown to constitute an atypical epithelium, with apical tight and adherens junctions and highly dynamic basal filopodia more typical of mesenchymal cells [43]. The epithelial properties of SHF cells are emerging as an important regulatory node controlling SHF deployment and are regulated by cell adhesion molecules and components of the planar cell polarity pathway, such as WNT5A and VANGL2, as well as TBX1, required for focal adhesion and filopodial activity in the SHF [33, 38, 43, 44]. Moreover, analysis of apical junctions, cell shape and actomyosin distribution in the SHF has suggested biomechanical models driving SHF deployment, including planar cell polarity driven push and pull mechanisms and an epithelial tension driven circuit promoting proliferation in the posterior region of the dorsal pericardial wall [45, 46].
Cardiac neural crest cells, like the SHF, move through the pharyngeal region into the developing arterial pole of the heart, playing an important role in dividing the embryonic outflow tract into the pulmonary trunk and ascending aorta [47]. Ablation of neural crest cells thus leads to persistent common arterial trunk due to a failure of outflow tract septation; cardiac neural crest cell ablation also leads to a shortened outflow tract through reduced SHF deployment (Figure 2A) [48, 49]. This observation in avian embryos was among the first to show the significance of SHF addition for cardiac looping and outflow tract alignment. These phenotypes reflect signal exchange between SHF and neural crest cells as they converge on the arterial pole of the heart, orchestrating normal outflow tract elongation. The T-box transcription factor TBX1 has been identified as a critical regulator of SHF development, promoting progenitor cell proliferation and delaying differentiation in the SHF [40]. TBX1 is the major gene implicated in 22q11.2 deletion or DiGeorge syndrome, characterized by craniofacial and cardiovascular defects, including a range of conotruncal CHD [50, 51]. Single-cell RNA-seq experiments have shown that Tbx1 controls myogenic trajectories from the multilineage primed progenitor cell population to cardiac and skeletal muscle fates [23]. Tbx1 null embryos display common arterial trunk associated with defective cardiac neural crest cell development; in addition, cells giving rise to myocardium at the base of the pulmonary trunk, the outlet of the right ventricle, fail to contribute to the arterial pole of Tbx1 mutant hearts [40, 52, 53]. Underdevelopment of subpulmonary myocardium has been proposed to be a primary upstream mechanism leading to tetralogy of Fallot [54]. Indeed, tetralogy of Fallot is genetically heterogenous but the major known genetic cause is haploinsufficiency for TBX1, 22q11.2 DS accounting for over 15% of patients [50]. Moreover, SHF ablation in avian embryos leads to a tetralogy of Fallot-like phenotype [55]. Distinct genetic programs have been defined in subpulmonary and subaortic myocardium, reflecting earlier differences in the distribution of progenitor cells in the SHF; recently Pparg and Dlk1 have been shown to demarcate subpulmonary and subaortic myocardium respectively [53, 56, 57]. Future subpulmonary myocardium, initially located in the inferior wall of the midgestation outflow tract, becomes positioned ventrally during outflow tract septation through counterclockwise rotation of the myocardial wall driven by ongoing SHF addition (Figure 2A) [58, 59].
Genetic studies have identified mutations associated with sporadic and familial CHD, including mutations in genes encoding T-box transcription factors and chromatin regulators [60]. Examples include TBX1 and CHD7, the latter implicated in cardiovascular defects associated with CHARGE syndrome. CHD7 is an ATP-dependent chromatin remodeler required for normal contributions of both neural crest and SHF lineages during heart development, including regulation of Fgf10 expression in the SHF through the ISL1-bound intronic enhancer [61]. Integration of genetic studies with single cell RNA-seq datasets can provide further mechanistic insights and has, for example, highlighted the importance of outflow tract progenitor cells in tetralogy of Fallot [62]. However, genetic defects only account for one third of CHD cases to date, suggesting an important, yet poorly understood, effect of in utero exposure to environmental factors. Indeed, recent studies in the mouse have identified the SHF as the target of short-term gestational hypoxia, maternal iron deficiency and prenatal alcohol exposure, in each case leading to reduced deployment of SHF progenitor cells and a shortened OFT, resulting in a range of conotruncal CHD [63, 64, 65].
4. TBX5 and cardiac septation
Dye labelling and genetic lineage tracing experiments have shown that subpulmonary myocardium originates in Hoxb1 expressing cells in the posterior lateral region of the dorsal pericardial wall, corresponding to the location of proliferative multilineage primed progenitor cells [23, 53, 66]. Downregulation of Hoxb1 expression is required for normal arterial pole and right ventricular development [67]. Cardiac progenitor cells in the Isl1, Hoxb1, Tbx1, Wnt5a and Mef2c enhancer genetic lineages contribute not only to the arterial pole but also to the venous pole of the heart, making critical contributions to atrial septal structures including the primary atrial septum and dorsal mesenchymal protrusion that bridges the atrial septum with the atrioventricular cushions (Figure 3A) [53, 67, 68, 69, 70]. Defects in dorsal mesenchymal protrusion development result in primum type atrial septal defects, a class of atrioventricular septal defect [71].
Venous pole development and atrial septation. (A) Scheme of the region boxed in Figure 2A, showing progenitor cell segregation into anterior SHF (aSHF) and posterior SHF (pSHF) domains contributing to the arterial and venous poles. Note the contribution of first heart field (FHF) cells from the juxtacardiac field (JCF) to the venous pole. (B) Diagram showing that Tbx1 expression is maintained in the aSHF, while Tbx5 is activated in the pSHF. (C) Left: the SHF genetic lineage (blue) labelled by a conditional β-galactosidase reporter gene activated by Cre recombinase in the SHF, showing labeling in the right ventricle (RV), interventricular septum (VS) and atrial septal structures: the primary atrial septum (PAS) and dorsal mesenchymal protrusion (DMP). Right: the DMP fails to form in Tbx1 mutant hearts resulting in a primum type atrial septal defect (asterisk). AP, arterial pole; VP, venous pole; A, atrium; V, ventricle; RV, right ventricle; LV, left ventricle; HT, heart tube. Scale bar in C: 100 μm
In contrast to the anterior SHF, which retains the early SHF genetic program, posterior SHF cells activate Tbx5, a key regulator of the FHF and venous pole differentiation program, under the influence of retinoic acid signaling [72, 73]. Tbx5 is upregulated in TBX1 positive pharyngeal mesoderm as the dorsal mesocardium breaks down and progenitor cells segregate to alternate poles (Figure 3B). Transient co-expression of TBX1 and TBX5 is followed by downregulation of the anterior SHF program and the establishment of a transcriptional boundary within the dorsal pericardial wall, separating TBX1 positive arterial pole progenitors in the anterior SHF from TBX5 positive venous pole progenitors in the medial posterior SHF [73]. Mutations in TBX5 result in Holt-Oram syndrome, characterized by atrial septal CHD and forelimb defects [74]. TBX5 has recently been shown to activate an enhancer of Aldh1a2, encoding a retinoic acid biosynthetic enzyme, pointing to a feed-forward mechanism between TBX5 and retinoic acid signaling as the posterior SHF domain is established in the dorsal pericardial wall [75]. The boundary between anterior and posterior domains is perturbed in the absence of Tbx1, resulting in failure of dorsal mesenchymal protrusion development and atrial septal defects in 30% of Tbx1 null embryos, potentially accounting for a low frequency of atrial septal defects in 22q11.2 DS patients (Figure 3C) [53]. Similar atrial septal defects are observed in conditional mutant mouse embryos lacking Tbx5 in the SHF [76]. The posterior SHF shares lineage contributions with pulmonary mesenchyme and signal exchange between the SHF and adjacent pulmonary endoderm plays an important role in venous pole development [75, 77]. TBX5 activates Wnt ligand expression in the posterior SHF that signals to pulmonary endoderm, in turn controlling the timing of differentiation in the posterior SHF through the Hedgehog signaling pathway [78, 79].
Cells expressing both Tbx5 and the SHF program have also been identified in the interventricular septal region using an intersectional lineage reporter approach [4]. Conditional inactivation using Cre recombinase driven by the Mef2c enhancer has shown that Tbx5 is required in the SHF lineage for formation of the muscular ventricular septum as well as for atrial septation [76, 80]; moreover, the transcription factors MEF2C and TBX5 coregulate target genes required for ventricular septal development [81]. Thus both atrial and ventricular septal structures form at sites where Tbx5 expression and the SHF program transiently overlap, suggesting that this defines a boundary program in the developing heart required to initiate septal morphogenesis. Atrial and ventricular CHD together account for over 50% of CHD [82]. Further dissection of this regulatory program, operating at the interface between the heart fields, will be essential to increase our understanding of the origins of septal CHD.
5. Conclusions and perspectives
Since the discovery of the SHF extensive evidence has revealed the importance of this spatiotemporally heterogenous progenitor cell population in the etiology of CHD affecting development of the cardiac poles and septa. While severe heart tube elongation defects result in early embryonic lethality, milder perturbation of cell addition at the cardiac poles results in alignment defects and failure to separate the systemic and pulmonary circulation at birth, contributing to a spectrum of common forms of CHD encountered in human patients. Technological advances, such as single-cell transcriptomics and quantitative morphometric approaches, as well as dynamic imaging and establishment of a variety of stem cell derived cardiac organoid models, are providing new insights into the regulation of SHF development [31, 83]. Current objectives include elucidation of the regulatory networks controlling distinct SHF subpopulations implicated in CHD, the identification of downstream pathways effecting regional heart morphogenesis and cardiac septation, and dissection of the disease mechanisms by which genetic and environmental factors interact in the SHF to cause CHD.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
Work in RK’s group is supported by the Agence National pour la Recherche (PP-heart ANR-20-CE13-0029-01 and Heartbound ANR-22-CE13 projects) and the AFM-Telethon. CG acknowledges the support of the French Government program managed by the French National Research Agency (ANR-16-CONV-0001) and the Excellence Initiative of Aix-Marseille University A∗MIDEX (Turing Centre for Living Systems) and the Fondation pour la Recherche Médicale. RK is an Inserm research fellow.