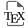
 CC-BY 4.0
CC-BY 4.0
It is now widely accepted that multicellular organisms surpass the concept of autonomous entities, evolving into intricate systems known as “holobionts” [1, 2]. These holobionts encompass not only the host organism but also a consortium of commensal and mutualistic microorganisms, alongside a diverse array of parasite taxa, including viruses, bacteria, fungi, protozoans, and metazoans. Remarkably, parasites themselves possess a microbiome, adding a layer of complexity to the interactions that take place [3]. Research has extensively illuminated the substantial influence of parasites on the phenotype of their hosts, uncovering interconnected eco-evolutionary dynamics between animals and symbionts. This interconnection manifests itself in virtually every facet of an animal’s behavior, influencing the likelihood of exposure to infection and subsequent alterations in infection consequences [4]. Furthermore, the behaviors of infected hosts can be affected by the presence of parasites, resulting in well-documented changes in host phenotype [5]. Notably, such alterations often prove advantageous for the transmission and survival of parasites or, conversely, represent adaptive responses by hosts aimed at eliminating infections or minimizing associated costs [6]. Recent studies have expanded this paradigm by demonstrating that microbes, particularly those constituting the intestinal microbiota, exhibit interactions with hosts akin to parasites. Lifestyle factors and social behaviors of hosts have been shown to influence the microbiota, and reciprocally, the intestinal microbiota can impact host physiology, mood, and behavior (e.g., [7]). These intricate relationships underscore a dynamic network shaping the biology and behavior of multicellular organisms, highlighting the multifaceted interplay between hosts, parasites, and microbiota.
Beyond microbiota and parasites, multicellular organisms share a complex evolutionary history with a distinct category of internal entities: malignant cells [8]. Originating from normal cells that undergo a shift in behavior during the host’s lifetime, becoming malignant, these cells proliferate at rates surpassing those of normal cells. Oncogenesis, which is omnipresent among multicellular species, occurred for nearly a billion years, dating back to the transition from unicellular to metazoan life (Appendix A). The evolution and diversification of multicellular organisms would have been unattainable without the establishment of diverse and efficient anticancer defenses (Appendix B), which indirectly underscores the importance of cancerous processes in the evolution of metazoans.
While not always leading to invasive cancers, there is a growing recognition that oncogenic phenomena, spanning from precancerous lesions to metastatic cancers, are prevalent in host populations [9]. This challenges the conventional perspective that primarily focuses on relatively well-developed tumors, which are most often found in post-reproductive individuals. Over the course of an organism’s life, the duration of interactions between a host and its oncogenic manifestations can vary from a few months to several years, or even several decades, with effects on the host that are not fully documented at all stages.
In contrast to microbiota and parasites, malignant cells typically do not spread between host individuals, except for some exceptions where cancer becomes transmissible, yielding the emergence of a novel (parasitic) species, which subsequently undergoes its own evolution (Appendix C).
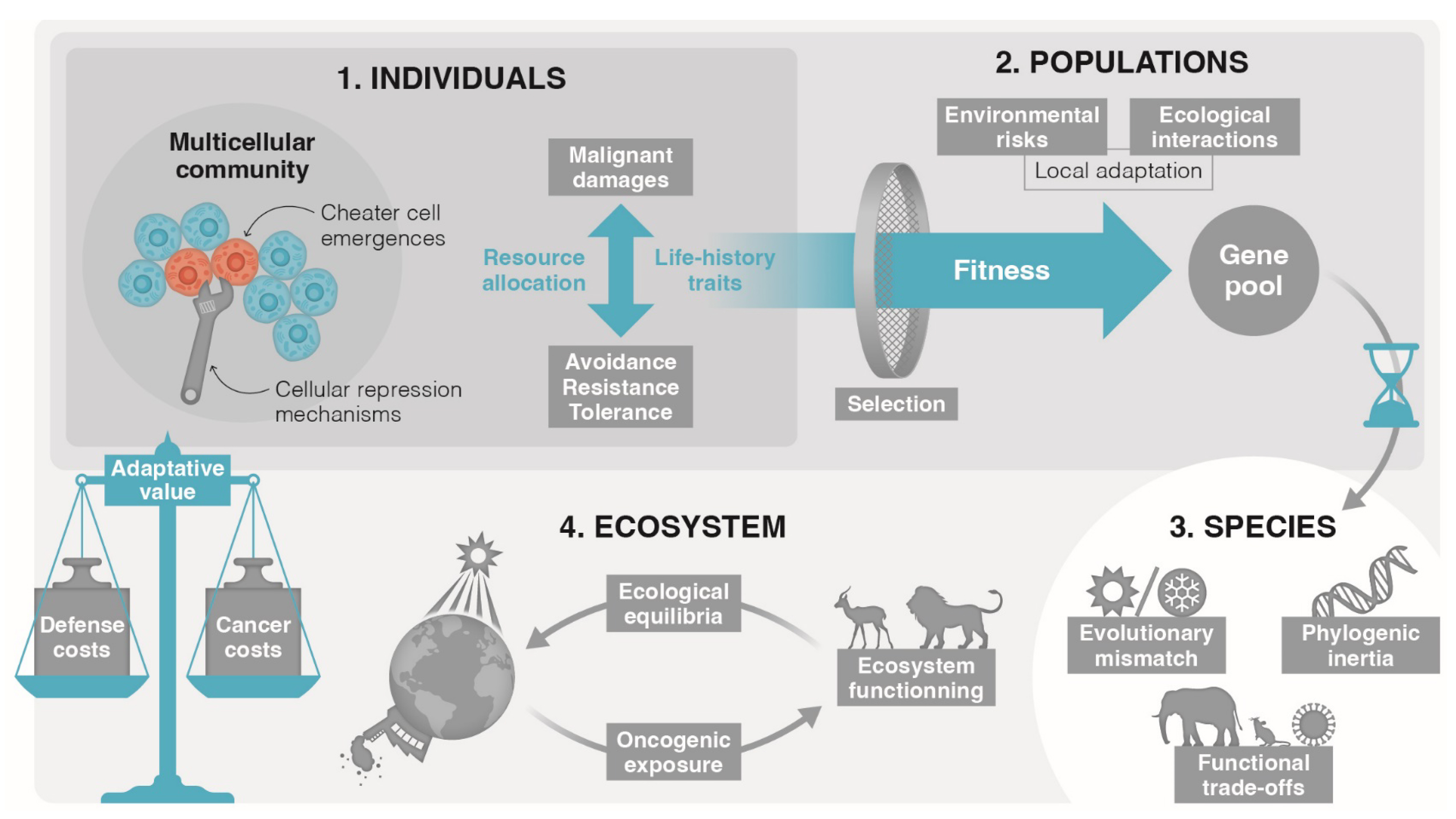
Akin to microbiota and parasites, specific host phenotypic traits can influence the dynamics of malignant cells, and these cells, in turn, can instigate phenotypic alterations in the host. Certain host phenotypic traits may act as underlying causative factors for cancer, while simultaneously being influenced by the progression of malignancy, potentially leading to intricate cycles reminiscent of those observed in specific host-parasite interactions [10]. Furthermore, mounting evidence suggests reciprocal interactions between malignant cells, symbiotic microbes and parasites, indirectly impacting the interplay between host phenotype and symbionts (Figure 1). The potential for triple reciprocal interactions involving malignant cells, symbionts, and hosts has been historically overlooked, primarily because evolutionary biologists have tended to disregard or classify as “noise” the host phenotypic variations resulting from oncogenic processes.
Interactions between oncogenic processes, host, infectious agents and microbiota (adapted from [10]).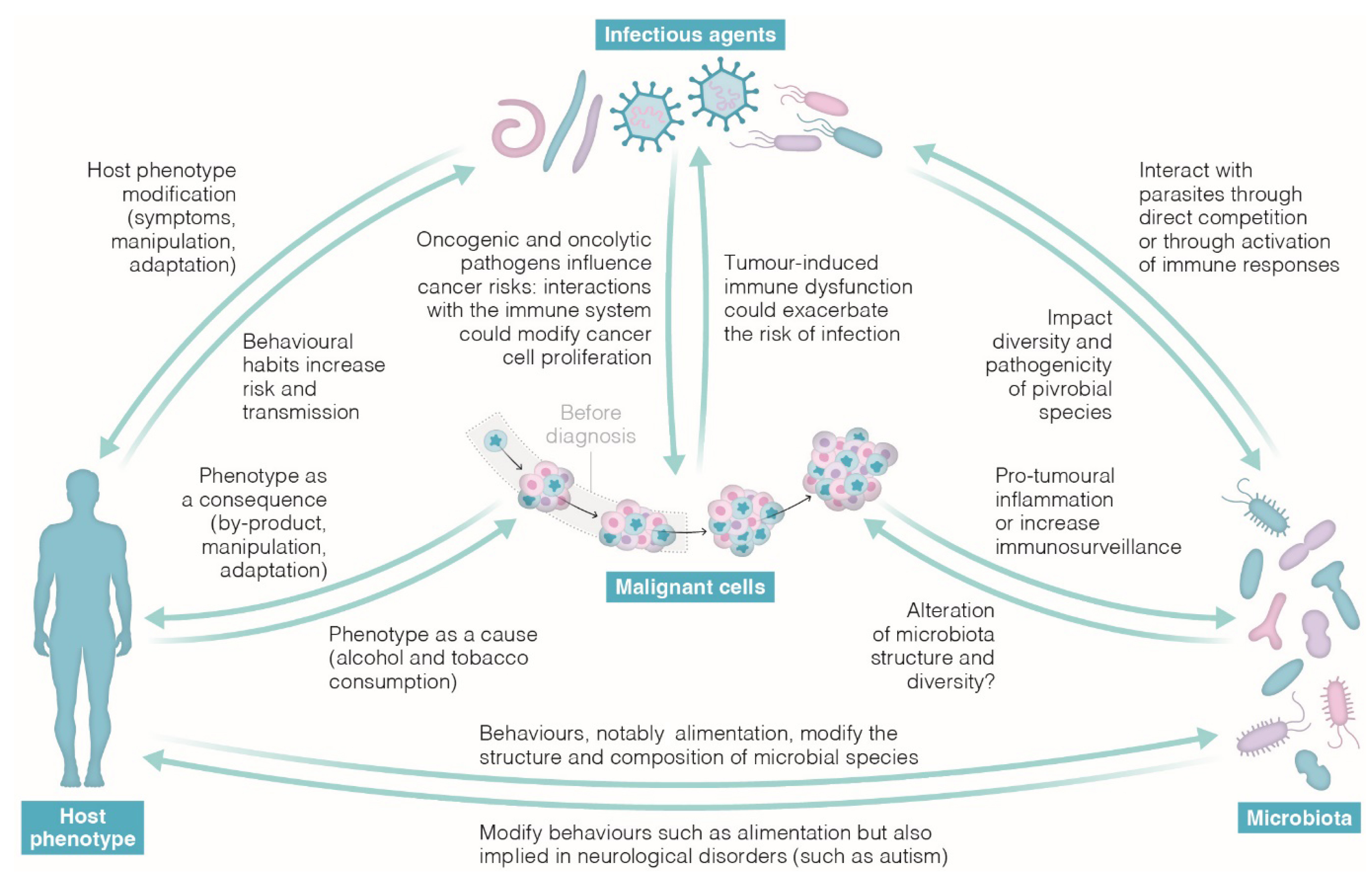
In this article, we delve into several critical dimensions of cancer biology and its ecological implications. First, we explore the profound impact of cancerous processes on host phenotype, shedding light on how subclinical tumor stages can contribute to broader interindividual variability. Next, we examine the intricate interactions between cancer and symbiotic organisms, unraveling their implications for cancer dynamics within ecological contexts. We then pivot to discuss the evolutionary ecology of host-tumor interactions in polluted ecosystems, highlighting how environmental factors shape oncogenic phenomena. By synthesizing these insights, we aim to elucidate the complex interplay between cancer biology, ecology, and evolutionary processes, offering a comprehensive perspective on the multifaceted nature of cancer in natural environments.
1. Effect of cancerous processes on phenotype
Most investigations into phenotypic changes in individuals with tumors have focused on post-diagnosis periods, typically when tumors become detectable, which may occur years or even decades after the onset of cancer. There is a need for a more comprehensive exploration of when phenotypic changes induced by malignant cells occur during the early stages of tumorigenesis, because it is plausible that the process itself triggers some alterations in the host phenotype. Given the parasitic-like interactions between malignant cells and their hosts, it is first reasonable to assume that these cells may impact the host phenotype through unintended consequences, such as non-functional by-products that are adaptive for neither party (see for instance [11]). Beyond these unintended effects, there is also an expectation that malignant cells might contribute to host manipulation [12], especially when tumors are transmissible [13]. Changes in the host phenotype may also arise from adaptive responses by the host aiming to eliminate the malignant cells (e.g., [14]). Cancer defenses designed to counteract malignant cells (see Appendix B), similar to general protective mechanisms, are likely to involve trade-offs with other functions with consequences at the cell, individual, population, species, and ecosystem levels (see [15, 16, 17] for reviews, Appendix D). Hence, even in seemingly cancer-free organisms, the influence of oncogenic processes on the host phenotype should not be disregarded, given the potential costs of preventing the progression of microscopic cancer cell colonies into lethal tumors. In cases where the growth of cancer is inevitable, individuals may react adaptively to minimize impacts, such as adjusting life-history decisions to reproduce early before the detrimental effects of cancer become pronounced (see Appendices B and D). Recent arguments (see Appendices B and D) suggest that phenotypic alterations beneficial for individuals with tumors, maximizing their fitness before death, may inadvertently support tumor progression (or transmission in the case of the tumoral hydra), especially if cancer-induced death occurs late in life when natural selection is weakened.
2. Interactions with symbiotic organisms and their implications in cancer dynamics
The role of infections in the genesis of cancer is well acknowledged, with an expanding list of pathogens identified as potential contributors to oncogenesis both in humans and wildlife [18, 19]. The immune system’s central role in surveilling and regulating malignant cells is crucial, and any disruptions in immune homeostasis, often triggered by infections, can impact the proliferation of malignant cells [20, 21]. An underexplored scenario we propose here is that oncogenic phenomena in wildlife may initiate complex cycles. Hosts grappling with poor health conditions due to malignancies may become more susceptible to increased parasite occurrences and infection intensities, further compromising their resilience and heightening the likelihood of subsequent infections and cancer progression. Given the ubiquitous presence of parasites, the coexistence of co-infections, and the persistent proliferation of malignant cells throughout an organism’s life, there is a pressing need for a deeper understanding of how parasite communities may either mitigate or exacerbate the processes of carcinogenesis[20, 21, 22].
In addition to potential interactions between malignancies and pathogens, there exists a compelling interplay with the microbiota, gaining recognition for its potential role in carcinogenesis [23, 24, 25]. Altered interactions among the microbiota, intestinal epithelium, and the host immune system have been associated with various diseases, including cancer [26, 27]. The microbiota’s pro-tumoral role, linked to inflammation, comes to the forefront, with disruptions in mucosal/epithelial barriers leading to the induction of pro-inflammatory cytokines, ultimately fostering tumor growth [27, 28, 29].
Colon carcinogenesis, as an illustrative example, is profoundly influenced by the microbiota inhabiting the colon. These microorganisms play a crucial role in various aspects of colon health, including the metabolism of substances like retinoic acid, which can impact cancer development [30]. Furthermore, the complex interactions between malignant cells and the microbiota extend to their influence on the host immune system. Malignant cells and their byproducts can affect the effectiveness of adaptive immunity, which in turn influences the diversity and abundance of the microbiota [31, 32]. Conversely, the microbiota itself plays a significant role in modulating immune responses against tumors, contributing to anti-tumoral surveillance mechanisms that help prevent or limit tumor growth [33]. This bidirectional relationship underscores the dynamic interplay between the microbiota, the immune system, and colon cancer progression.
Microbial products further mold T-cell repertoires and may regulate anti-tumor responses by priming cross-reactive T cells specific to both bacterial and tumor antigens. The exploration of host-microbiota interactions and the determination of specific microbiota’s causal roles in cancer remain vibrant areas of research. While the presence of bacteria in tumors may hint at associations with certain cancers, it could also signify local infections within existing malignant tissue, resembling opportunistic infections within established tumors [33]. Beyond bacteria, there is growing evidence supporting the role of both commensal and pathogenic fungi in influencing various cancer-related processes. These mechanisms encompass local effects within the tumor microenvironment and remote influences through the secretion of bioactive metabolites, modulation of host immunity, and interactions with adjacent bacterial commensals [34, 35]. However, it is a recent field of research still in its infancy, and much is still to be uncovered, especially in wildlife species.
3. Ecology and evolutionary consequences of oncogenic phenomena
The presence of oncogenic phenomena in hosts is anticipated to induce significant changes in key ecological variables, encompassing competitive abilities, foraging strategies, metabolism, immune competence, vulnerability to predators, and dispersal capabilities [36]. These anticipated effects parallel our expectations for how microbiota and parasites influence these ecological processes [37]. However, the indirect impacts on ecosystem functioning and the potential evolutionary feedbacks, where the host’s biology is shaped by oncogenic processes, remain poorly understood at the moment. The consequences of varying susceptibilities to cancer among species will depend on the ecological status of the most impacted species, if they are for instance predators, prey, or keystone species [38, 39, 40]. For instance, transmissible cancer in Tasmanian devils (Sarcophilus harrisii) has significantly reduced the population size of this scavenger species, leading to a series of cascading effects with major ecological consequences, including impacts on the abundance of invasive species [41]. Let’s, however, keep in mind that the dramatic consequences observed at the ecosystem level with the Tasmanian devil’s cancer are undeniably linked to the fact that this cancer is transmissible, which means it can cause the same devastation as a other infectious agent. In other words, the consequences at the ecosystem level with classic, non-transmissible cancers are certainly less severe. The phenotypic changes induced by oncogenic processes in hosts also have the potential to alter various interactions between the affected organisms and other species within the ecosystem. For example, tumor-bearing hydra exhibit modified interactions with other species compared to their healthy counterparts—they capture more prey, are more susceptible to predation, and experience heavier colonization by commensal ciliates [42] (see also [43]). Recently, Tissot et al. [44] also experimentally demonstrated that tumoral hydra act as superspreaders of commensal ciliates, i.e., not only do ciliates multiply faster on tumoral polyps, but this phenomenon also leads to a higher colonization rate on other hydra polyps, whether they are of the same species or not (i.e., spillover). In general, it is known that cancers often lead to immunosuppression in hosts, and consequently it is important, from both a health and conservation perspective, to assess how the resurgence of cancerous pathologies in wild animals may boost parasitic communities and their circulation.
In instances where the changes triggered by malignant cells in host phenotypes occur before the end of the reproductive period, their influence could extend to immediate and long-term natural selection pressures on hosts. Consequently, selection becomes shortsighted when the correspondence between genotype and phenotype is reduced. Analogous to the effects observed with microbiota and parasites (e.g., [45]), malignant cells may disrupt the linkage between selection acting on phenotypes and selection operating on genotypes. Consequently, a plausible outcome of the influence of malignant cells could be a deceleration of evolutionary changes, particularly as long as individuals with tumors persist in passing on their genes to successive generations.
4. Evolutionary ecology of host tumor interactions in polluted ecosystems
A growing body of research indicates that wildlife is increasingly susceptible to cancer compared to the past [46, 47, 48]. Examples in emblematic species include St. Lawrence beluga whales (Delphinapterus leucas), green sea turtles (Chelonia mydas), California sea lions (Zalophus californianus), and numerous fish species. The most plausible hypothesis for this abnormal increase in cancers is attributed to human activity, leading to humans being labeled as the “oncogenic species” for wildlife [47]. As for humans, the escalating oncogenic risks produced by anthropic activities render previously evolved anticancer defenses inadequate (i.e., evolutionary mismatch, see Appendix D). These activities encompass mutagenic pollution of the environment, dietary changes, and reductions in genetic diversity (intensifying inbreeding issues) of wildlife species. Exposure to mutagenic sources can influence various genetic and physiological processes in organisms, requiring a comprehensive understanding of their collective contribution to the observed phenotypic changes in each species. In the context of conservation facing this unprecedented situation, it is crucial to move beyond more observations and precisely dissect the diversity and impact of all underlying processes [49]. To achieve this, it is essential to comprehend the short-, medium-, and long-term phenomena observed in organisms and species suddenly exposed to an oncogenic environment surpassing the one in which their current anticancer defenses evolved (Figure 2). These encompass: (i) the expenditure associated with activating anticancer defenses (and the associated side effects); (ii) the costs of tumorigenesis itself; (iii) alterations in life-history traits; and (iv) the selection for enhanced anticancer mechanisms. The potential new evolutionary paths that wildlife species may take will be shaped by the interplay of these effects with additional selective pressures imposed by the biotic and abiotic conditions within ecosystems.
Short, medium, and long-term effects of wildlife exposure to mutagenic ecosystems (adapted from Dujon et al. [49]).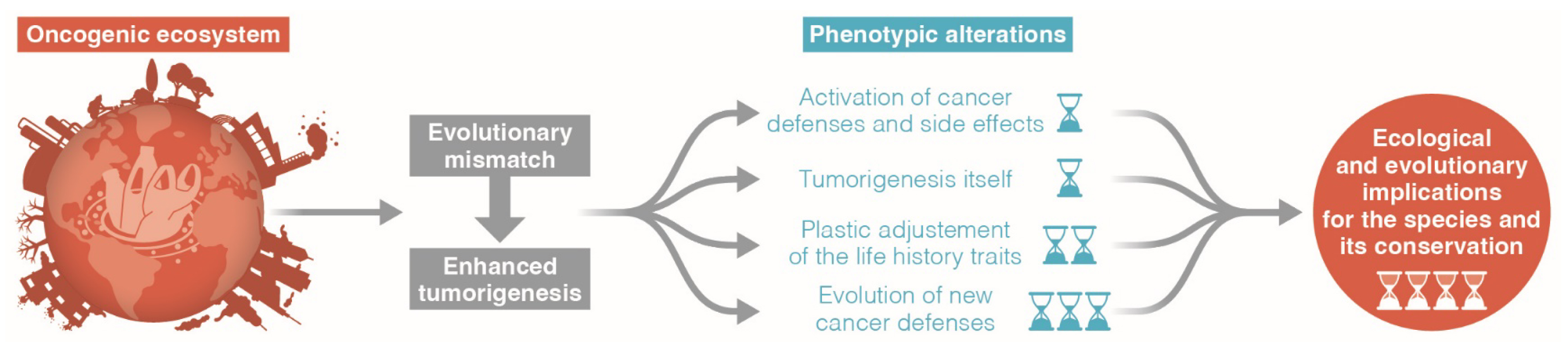
A first aspect to consider is the expenditure associated with triggering anticancer defenses. As seen in Appendix B, cancer defenses are various, but irrespective of the specific defense mechanisms mobilized, none of them come without associated costs; they all consume energy that could otherwise be allocated to other bodily functions, giving rise to trade-offs. In an environment undergoing rapid and abnormal mutagenic changes, the prevalence of oncogenic processes may significantly rise, prompting the consistent activation of defenses aimed at thwarting them. The escalated cost associated with this systematic activation is anticipated to disrupt evolutionary trade-offs and impose substantial adverse effects on traits such as survival and/or reproduction (see Appendix D). It is therefore possible to consider that these defense mechanisms will be, nowadays, a sort of Darwinian trap with ultimately negative effects on fitness. This research field remains, at the moment, poorly explored but appears highly relevant. Accordingly, some recent studies also suggest that the cost of activating anticancer defenses is indeed capable of altering the biology of species (e.g., [50]). It is interesting to note that this hypothesis also leads to the counterintuitive situation in which we do not observe cancer in a mutagenic environment (due to the effectiveness of defenses), even though organisms will have their biology significantly altered due to trade-offs. These considerations are undoubtedly crucial in terms of conservation.
Another important aspect to consider for a better understanding of the factors influencing phenotypic variations in organisms in mutagenic environments is, of course, tumorigenesis itself. Organisms will host various stages of cancerous processes, more or less advanced, and these processes are not neutral regarding body condition. On the medium term, selection is expected to adjust life-history traits (see examples in Appendix B). Additional research is required to investigate the extent to which adjustments in life history traits favor population-level and individual responses to cancer risks, and how easily or rapidly this occurs. If organisms remain exposed to mutagenic environmental factors over extended periods, natural selection may favor individuals possessing stronger anticancer defenses, conferring a selective advantage. However, it is important to consider that the enhancement of anticancer defenses through selection depends on specific genetic variations, which may not always occur (given that mutations happen randomly, limiting the emergence of “beneficial mutations”). When such selection is hindered, it is anticipated that other compensatory adaptations (e.g., menopause as proposed before [51]) will be favored, at least temporarily, as alternatives.
Thus, several processes can induce phenotypic changes in organisms experiencing an evolutionary mismatch concerning cancer vulnerability. Tasmanian devils exhibit resistance, tolerance, or adjustments in life-history traits in response to the transmissible cancer DFTD. A critical question is whether adjusting life-history traits is easier to select for than improved cancer resistance. As oncogenic pressure intensifies, relying solely on life history trait adjustments may not suffice for long-term species preservation, necessitating the development of superior anticancer defenses. For example, in DFTD-infected Tasmanian devil populations, individuals reproduce sooner, but further delay reduction is constrained by seasonality. The transition from life-history trait adjustments to resistance selection might be occurring, emphasizing the need for continued investigation. This perspective holds significance not only in conservation biology but also in ecology and evolutionary biology. Inter-individual variability, traditionally attributed to genetics or factors like parasitism, may partly result from subclinical tumor stages. The consequences of this variability, particularly in early tumorigenesis stages, and its interaction with anticancer defenses activation remain poorly understood. Ecological status influences species’ persistence, with potential cascading effects and complex dynamics. The evolving interplay between host-tumor interactions in mutagenic environments demands thorough exploration, offering insights into cancer’s role in the evolutionary history of multicellular organisms.
5. Concluding remarks
The phenotypic variations between individuals with tumors and their healthy counterparts largely result from the complex interaction between a tumor and its host. Exploring these phenomena in the wild is of crucial importance to deepen our understanding of the pivotal role played by oncogenic processes in the evolutionary ecology of hosts and the functioning of ecosystems. Historically, ecologists overlooked the roles of parasites and the microbiota in host biology and evolution. However, with the use of more refined methodologies, it becomes evident that neglecting these aspects is no longer tenable. We advocate for the recognition of the importance of malignant cells in evolutionary biology and ecology, emphasizing their equivalent significance with parasites and the microbiota (see Figure 1). To embark on this innovative approach, experimental research should initially leverage practical animal model systems such as zebrafish, mice or drosophila. This approach would allow the manipulation of the microbiota, controlled initiation of cancer (e.g., through genetically modified organisms), and observation of host phenotypic changes from the onset of cancer to the conclusion of the life cycle. A multidisciplinary approach, including mathematical and computational modeling, will play a central role in developing a holistic perspective. We hope that the concepts addressed in this paper will consolidate the foundation for a thorough exploration of the underestimated role of oncogenic processes in the biology, ecology, and evolution of multicellular hosts. Finally, we have repeatedly highlighted in this article the impact of cancerous processes on host phenotypic variation, noting for instance that subclinical tumor stages could contribute to interindividual variability otherwise attributed to genetics or parasitism (see above). We propose to strengthen studies considering cancerous processes as a potential outcome of embryonic development, thus suggesting an integration of cancer into the developmental mechanisms that influence the genotype-phenotype map. This approach could enrich the field by integrating concepts from evo-devo to better understand these complex dynamics [52, 53, 54].
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was supported by the EVOSEXCAN ANR project (ANR-23-CE13-0007) and the Hoffmann Family.
Appendix A. Cancer, an evolutionary phenomenon that emerged with the advent of multicellular organisms
Where does cancer originate? The biological process that leads to this disease is found in all vertebrate classes, including emblematic extinct species like the Jurassic dinosaurs [10, 55, 56]. Cancer is also observed in invertebrates such as insects, mollusks, and cnidarians, like hydras [57, 58]. In actuality, the inception of cancer dates back approximately one billion years, coinciding with the emergence of multicellular organisms in the late Precambrian era [59] (the oldest known cancer fossils date back 240 million years in turtles [60], and 1.7 million years in humans [61]). During this period, life on Earth evolved from unicellular organisms to more intricate entities known as metazoans, consisting of thousands, or even billions, of genetically identical cells working collaboratively. To achieve this, these cells activate various segments of their genome to specialize and cooperate through task allocation [62]. Once this effective cooperative functioning logic was established, natural selection favored the most successful cooperative entities—those capable of maximizing their reproductive success. This involved excelling in resource acquisition, avoiding predators and parasites, attracting sexual partners, etc. However, paramount was the need to maintain the organism’s functionality. To achieve this, natural selection during the evolution of multicellular organisms favored adaptations that strengthened cohesion and communication between cells to harmonize their collective functioning. Simultaneously, other adaptations aimed to prevent, control, or eradicate cells that did not contribute to this collective effort, thereby restoring unicellular functioning. These adaptations, selected over millions of years, have significantly contributed to the remarkable diversification of multicellular organisms observed today.
The benefit of this collective operation lies in its formidable efficiency compared to unicellularity. However, like all collective systems, it remains vulnerable to cheaters. Cheating occurs when someone gains the benefits of a cooperative system without fulfilling their necessary contribution to its proper functioning. Thus, one of the primary challenges in the evolution towards functional multicellularity has been constraining some of the constitutive cells to give up their own ability to reproduce. This represents a significant sacrifice in Darwinian terms, as natural selection typically favors maximizing reproduction. Nonetheless, the logic holds because somatic cells, which are non-reproductive, still indirectly promote the transmission of their genes through the reproductive sex cells, with which they share the same genome.
Occasionally, following mutations, certain somatic cells, despite their non-reproductive nature, attempt reproduction nonetheless. If the resulting conflict of interest is not resolved, it can lead to tumor formation and eventually cancer. As this challenge has persisted since the emergence of multicellularity, selection has favored the evolution of anticancer adaptations capable of effectively suppressing these cellular rebellions from that early period onwards [63]. Nevertheless, cancer cells represent living entities subject to an evolutionary process guided by natural selection. Consequently, they possess the ability to dynamically acquire adaptations in real-time, enabling them to evade anticancer defenses. This dynamic unfolds precisely during the progression of cancer. Ironically, it is the same natural selection process that, on one hand, fosters the development of defenses against cancer and, on the other hand, propels the disease to advance into an invasive state. Natural selection favors entities that are most effective at reproduction, irrespective of whether they confer benefits or harm. The shift from unicellularity to multicellularity throughout the history of life occurred not as a singular event but rather took place around a dozen times since the Precambrian era. The advantages associated with multicellularity played a pivotal role in driving this transition. However, in each instance, the social dilemma mentioned earlier emerged, contributing to the ubiquity of cancer as a disease within all multicellular groups [56].
Although cancer is an ancient ailment, it presents a unique narrative with each occurrence. Strikingly, in the majority of instances, cancer is not communicable (although there are exceptions, see Appendix C). As a result, the entire spectrum of genetic, epigenetic, and cytogenetic diversity established during tumorigenesis vanishes with the demise of the host, be it due to cancer or other causes. Consequently, cancer is compelled to innovate anew on each occasion [64]. It is noteworthy, given these circumstances, that with each occurrence, there is a transition from a cell detaching from the collective system to an intricate cooperative system—an ecosystem within the larger ecosystem (the solid tumor). This pattern exhibits profound resemblances between organs, individuals, and even species. Evolutionarily, potential explanations for this phenomenon include evolutionary convergences (where only cancer cells capable of organizing in this manner can elude anticancer defenses) [65], or the reactivation of ancestral genetic programs dating back to the Precambrian era (the atavistic hypothesis of cancer) [66, 67]. Recently, Thomas et al. [68] argued that it also represents a compelling example of selection for function (sensu [69]): Natural selection among individual cancer cells and cell groups within the tumor, which is originally the engine of tumor initiation, later serves as a tool to generate various configurations suitable for various changing and challenging environments, which is the key to selection for functional optimization and tumor progression.
Appendix B. Evolution of anticancer defenses among animals
Throughout metazoan evolution, the process of selection has endorsed the development of a repertoire of mechanisms designed to thwart the emergence of antisocial cells or identify and repress them [70]. These mechanisms function across various levels, encompassing genomes, cells, tissues, and the entire organism. Multicellular organisms have faced the selective pressures of natural evolution since their inception to combat cancer cells. In contrast, cancer cells have a relatively brief evolutionary history, with a lifespan typically limited to a few years. Consequently, host organisms have developed highly effective mechanisms to counteract cancer cells, given their longstanding adaptation to confront a recurring adversary with each new generation [10]. As a result, multicellular organisms, for the most part, experience invasive lethal cancers infrequently, particularly during their reproductive phase. However, post-reproductive stages witness a higher incidence of cancers, as natural selection no longer upholds the efficiency of anticancer defenses as robustly [71]. The logical extension of this reasoning is that elevating cancer risks, such as through exposure to an oncogenic environment (see after), leads to a situation where the anticancer defenses established through evolution are no longer optimally aligned with the increased risk [72, 73]. Invasive cancers are more prevalent than anticipated, exemplified by occurrences in domesticated canids and humans, who have undergone rapid and recent changes compared to their close ancestors [74].
Within the established framework of ecology and evolution, an increasing number of researchers have recently directed their focus toward exploring anticancer defense mechanisms within the animal kingdom. It is noteworthy that, despite the high prevalence of oncogenic processes in multicellular organisms, there are significant variations between species. This suggests, albeit indirectly, that certain species hold unique “anticancer secrets”, enabling them to more effectively prevent, eradicate, or tolerate tumors—reasons for which are still under investigation.
B.1. Peto’s paradox
Research into cancer resistance in the animal kingdom centers on the concept known as “Peto’s Paradox”. The underlying rationale is quite simple: if the initiation of cancer is primarily dependent on malfunctioning cells, then, logically, a large organism composed of numerous cells (such as a blue whale, Balaenoptera musculus, with seven million times more cells than a house mouse, Mus musculus domesticus), and boasting a lengthy lifespan (whales can live close to a hundred years, while domestic mice have a lifespan of three to four years), should be more susceptible to cancer. Each cell division, after all, comes with a risk of mutations leading to a malignant lineage. Within the same species, this prediction is often borne out: larger individuals generally exhibit a higher vulnerability to cancer than their smaller counterparts. Research in humans and dogs has shown that cancer risks are frequently positively linked with size [75]. However, this correlation appears to diminish when examining different species. Studies reveal no clear connection between cancer occurrence, body size, and lifespan. Hence, despite their imposing stature, whales and elephants do not demonstrate an elevated prevalence of tumors, suggesting an implicit resilience to cancer [76]. This paradox is once again elucidated through the lens of the evolutionary process. Large-sized and long-lived species have managed to endure and persist to the present by evolving robust cancer defense mechanisms through natural selection. To envision a scenario where whales, with a thousand times more cells than humans, experience a thousand times more cases of cancer suggests that they likely would not have survived.
To date, the most extensive examination of Peto’s paradox has been undertaken by Vincze et al. [77]. This study focused on captive wildlife in zoos, specifically looking at 191 mammal species where health data, including cancer information, had been systematically gathered (utilizing the Species360 database). Adjusting for various potentially confounding factors related to zoo conditions, the primary insights derived from this research are as follows: (1) Cancer-related mortality risk exhibits significant variation among species, ranging from 0% to 57.4%, as observed in the Kowari, a small carnivorous marsupial native to Australia; (2) The risk of cancer-related mortality surpasses 10% in 41 species, emphasizing that cancer is not confined to humans and their domesticated animals: it can affect a diverse array of species provided they live long enough, a condition often met in zoos; (3) A notable phylogenetic pattern is observed: carnivores tend to have a higher incidence of cancers compared to primates or artiodactyls, even when considering that the latter include large-sized species; (4) Addressing Peto’s paradox, the study affirms the lack of a straightforward correlation between size, longevity, and cancer risks.
More recently, Dujon et al. [78] have proposed the idea that the principles of Peto’s paradox might extend beyond size and longevity to encompass other traits. If a particular trait is expected to predispose to cancer but does not, there is a likelihood that natural selection has played a role in this divergence from the predicted outcome. An illustrative example is found in the various types of placentation among mammals, differing in invasiveness. While one might anticipate species with highly invasive placentation (hemochorial placenta) to be more susceptible to metastatic cancers, analysis of the same dataset on captive wildlife reveals this expectation does not hold true. In essence, species retaining invasive placentation have concurrently developed mechanisms that mitigate the risks of metastatic cancers.
B.2. What mechanisms are at play in preventing cancer?
In recent years, diverse studies have explored various species for their resistance to cancer, including naked mole-rats, elephants, whales, Placozoans, Xenarthrans (sloths, armadillos, and anteaters), and bats. The primary investigations into naked mole-rats were led by the team of Vera Gorbunova and Andrei Seluanov [79, 80, 81]. The naked mole-rats are widely recognized for their remarkable resistance to cancer, making them a focal point in oncology research. Several biological traits in these animals may contribute to their ability to prevent tumor development. Notably, fibroblasts in naked mole-rats have demonstrated the presence of two independent mechanisms that inhibit cell contact. These mechanisms involve specific regulatory pathways such as p53, pRb, p16, and p27, which could play a pivotal role in preventing cancer. The same authors also show that in hyperplastic precancerous cells, the loss of methylation induces the transcription of endogenous retroviruses and cell death through the interferon pathway [82]. Additionally, naked mole-rats exhibit adaptations in oxidative metabolism, favoring glucose utilization over oxidative phosphorylation. This metabolic preference potentially reduces oxidative stress, limits DNA damage, and contributes to their resistance to cancer.
The extended lifespan of naked mole-rats (up to 30 years) compared to similar-sized rodents is noteworthy, suggesting potential connections to more efficient DNA repair mechanisms or heightened resistance to cellular aging. Researchers have also explored the unique viscosity of cell cultures in naked mole-rats, attributed to a dense hyaluronic acid produced by the cells themselves and expelled into the extracellular matrix. This characteristic prevents cells from clustering and forming tumors. To validate whether this specific hyaluronic acid is indeed responsible for cancer resistance in naked mole-rats, researchers conducted experiments to block its production in 80 individuals. The results indicated a tendency for cells to cluster, raising the possibility of potential tumor formation (see however [83]). As suggested by the authors, it remains possible that the primary driver behind the selection for producing a distinctive hyaluronic acid might not be primarily for cancer protection but rather as an adaptation to the subterranean environment. The gradual development of remarkably flexible integument, serving to shield their exposed skin from injuries while navigating narrow tunnels dug with their teeth, could be the main purpose. In this scenario, protection against cancers might emerge as a beneficial side effect of another adaptation.
Several other mole-rat species, such as the Golan Heights blind mole-rat (Spalax golani) and the blind mole-rat from the Judean Mountains (Spalax judaei), display strong resistance to cancer. Initial expectations of finding similar defense mechanisms as observed in the naked mole-rat were not met. Researchers isolated cells from these rodents, cultured them, inducing multiplication beyond what naturally occurs in the animals’ organs. During the first seven to twenty phases of multiplication, everything proceeded normally. However, beyond the twentieth multiplication, the cells started to undergo rapid mortality [84]. Analysis of the dying cells revealed the initiation of interferon beta (IFN-β) release, triggering massive death of necrotic cells within three days. Essentially, once the cells recognized they had surpassed a critical multiplication threshold, apoptosis was activated. Notably, different proximal mechanisms have been selected in all cases to confer resistance to cancer processes. This underscores an evolutionary convergence where similar ecological contexts lead to analogous adaptations, even if not rooted in the same precursors.
The ability of elephants to resist cancer has been linked to specific genetic characteristics, particularly the amplification of the TP53 gene copies. The TP53 gene plays a crucial role in suppressing tumors and repairing damaged DNA. African and Asian elephants have been observed to possess multiple copies of this gene (around twenty), a notable contrast to humans, who have only one copy. When DNA damage occurs, the TP53 gene becomes activated, regulating the cellular response and potentially inducing programmed cell death (apoptosis) in damaged cells, thereby preventing tumor development. However, the precise functionality of these additional copies remains a topic of ongoing debate [85]. Another process acting in elephants is the reactivation of a “zombie gene” known as LIF6 [86]. While the LIF gene, a leukemia inhibitory factor, is typically present in a single copy in mammalian genomes, elephants have ten copies. However, aside from LIF6, these extra copies are non-functional, having accumulated mutations over time, rendering them incapable of producing their protein correctly—referred to as “pseudogenes”. In contrast, the LIF6 gene has regained functionality, actively contributing to the destruction of cancer cells. Importantly, the reawakened activity of this gene remains under the control of the TP53 gene. Thus, TP53 not only identifies cells with damaged DNA but also triggers other genes that reinforce anticancer defenses.
In Xenarthrans, a group that includes sloths, armadillos, and anteaters, various lineages have independently developed large body sizes, long lifespans, and an inherently reduced susceptibility to cancer. Recent investigations [87] demonstrate that this diminished vulnerability aligns with surges in duplications of tumor suppressor genes within the fundamental lineages of Xenarthrans and pilosids (pilosids are a subgroup of Xenarthrans, and they include anteaters and sloths, while Xenarthrans also encompass cingulata, such as armadillos). Sloths exhibit an exceptionally sluggish cell proliferation rate, while other Xenarthrans trigger apoptosis even at minimal doses of DNA-damaging agents. Consequently, the occurrence of cancer is remarkably low in Xenarthrans, with nearly no instances reported in armadillos. These findings imply that the amplification of tumor suppressor genes has played a pivotal role in the evolution of significantly large body sizes and a reduced risk of cancer in Xenarthrans, establishing them as remarkable cancer-resistant mammals.
In 2021, a study [82] exposed the Placozoan Trichoplax adhaerens to elevated doses of X-rays to explore how this species managed the presence of mutated cells. T. adhaerens displayed an impressive capacity to endure high levels of radiation, showcasing resilience in the face of DNA damage [82]. Upon exposure to X-rays, these organisms responded by expelling clusters of cells, ultimately leading to their demise. Essentially, they expelled potentially hazardous cells from their organism. This exposure to radiation also triggered heightened activity in genes associated with DNA repair and the apoptosis process.
Studies on bats suggest that their resistance to cancer can be attributed to at least two distinct factors: a reduction in the signaling of growth hormone and insulin-like growth factor-1 (IGF-1), coupled with alterations in microRNA patterns [81]. Both growth hormone and IGF-1 are pivotal molecules in cell growth and metabolic regulation, with heightened levels typically linked to tumor growth and cancer development. The diminished signaling of these hormones in bats may thus play a role in impeding tumor growth. MicroRNAs, small RNA molecules governing gene expression, undergo specific changes in bats that likely contribute to their cancer resistance. In the genomic analysis of the Mesoamerican mustached bat (Pteronotus mesoamericanus), researchers identified positive selection in 33 tumor suppressor genes and 6 DNA repair genes, potentially influencing their lower cancer susceptibility and increased lifespan [88].
Cetaceans, encompassing various species renowned for their size, such as the blue whale (Balaenoptera musculus) with an average length of 25 to 27 meters and weighing 130 tons, as well as their impressive longevity, exemplified by the bowhead whale (Balaena mysticetus) that can live over 200 years, have been the focus of extensive research. These studies have not consistently uncovered the presence of additional copies of tumor suppressor genes, a characteristic that seems to be lacking in certain species like the minke whale (Balaenoptera acutorostrata) [89], the bowhead whale (Balaena mysticetus) [32], the sperm whale (Physeter macrocephalus) [90], or the humpback whale (Megaptera novaeangliae) [91]. According to a study conducted by Sun et al. [92] encompassing 22 cetacean species, genes exhibiting an evolution correlated with body size are primarily associated with immune surveillance, tumor suppression, and the development of hypertumors (discussed later). Concerning the immune system, the authors propose that the primary focus of selection may not necessarily be on defending against cancer processes but rather on the necessity to combat parasitic infections. In larger species, which inhabit a broader range of habitats encompassing both freshwater and saltwater environments, parasitic infections tend to be more prevalent. These infections can manifest at an early age and pose a short-term threat, in contrast to cancers, which are seldom immediately life-threatening. Consequently, the evolutionary imperative might be centered around managing parasitic challenges, and the enhanced resistance to cancer could be an incidental outcome of other adaptive mechanisms. The concept of hypertumors is relatively obscure and original. Initially introduced in 2007 [93], this hypothesis is built on the notion that a strategy to combat tumors involves allowing other tumors to establish themselves within, leveraging the familiar principle that “the enemy of my enemy is my friend”. In contrast to the Peto’s paradox logic, which proposes that cancer resistance rises with size and/or longevity, the hypertumor hypothesis suggests that beyond a certain size/longevity threshold, it could be evolutionarily advantageous to deactivate specific anticancer defenses. Consequently, cancer cells, as exploiters, might exploit the cancer system they originate from, ultimately weakening the original tumor.
Finally, with the increasing availability of species genomes in scientific literature, comparative genomics is emerging as a valuable tool for exploring the evolution of anticancer defenses across the animal kingdom. This includes investigating instances of evolutionary convergence, as demonstrated in studies such as [94, 95, 96]. For instance, a comprehensive analysis [95], covering 193 vertebrates, spanning mammals, birds, fishes, and reptiles, indicated that genes exhibiting greater conservation in species with higher cancer rates were primarily associated with metabolic functions. Conversely, genes showing higher conservation in species with lower cancer rates were linked to cell cycle regulation, DNA repair, and immune response. These comparative approaches further support the notion that animal species with larger body masses have evolved specific cellular mechanisms to counteract mutation accumulation and maintain genetic stability. This adaptive trait may elucidate the absence of an increased incidence of cancer despite prolonged exposure to potential mutagenic factors. In summary, it is imperative to acknowledge the varied susceptibility of species to cancer. Contrary to expectations, larger and/or longer-lived species do not display higher susceptibility to cancers, and diverse solutions to Peto’s paradox have been identified (also refer to reviews: [97, 98]). This observation aligns with the fact that large sizes have independently evolved multiple times in the history of multicellular organisms.
B.3. Considering all ecological constraints
To gain a comprehensive understanding of the evolution of anticancer defenses in the animal kingdom, it is crucial to consider all the ecological constraints that animals face in real ecosystems. Anticancer defenses, by their nature, can only be optimal rather than maximal in the living world. This is because they are not the sole priority for organisms, and there are inherent compromises and constraints. For instance, in ecologically unstable environments, positive selection might have occurred for an increased germline mutation rate, promoting greater genetic diversity as part of a “bet-hedging” strategy, even though it could subsequently elevate cancer risks [99]. Furthermore, natural selection predominantly favors reproduction over survival. This means that strategies leading to improved reproduction will be favored, even if they entail an increased risk of cancer later in life [16]. In Tasmanian devils (Sarcophilus harrisii), engaging in fights and bites to access mates exposes individuals to the risk of contracting the transmissible and lethal cancer DFTD. However, a strategy of avoiding aggressive interactions with other individuals is not evolutionarily viable. Even if individuals adopting such a strategy were to live longer by avoiding DFTD, they would not pass on their genes to the next generation. From a natural selection standpoint, it is more advantageous to compete for reproduction, even if it comes with the risk of earlier death from cancer. Another consideration is that anticancer adaptations that are “too” effective might result in significant collateral damage, such as autoimmune diseases. This evolutionarily unsustainable constraint has likely been a significant obstacle to the potential selection of more potent anticancer adaptations. Additionally, certain adaptations (e.g., for rapid early-life growth, increased aggression) may rely on genetic variants associated with antagonistic pleiotropy, meaning they are retained by natural selection due to the advantages they provide early in life, even if they promote cancer development later on [100]. Natural selection has favored behavioral strategies in the animal kingdom to prevent or mitigate the impact of cancer. For instance, birds like tits and flycatchers around Chernobyl can choose nests in less irradiated areas [101]. In the case of drosophila with cancer, they tend to prefer social environments that slow down the disease’s progression [102]. Additionally, research in various species, including fruit flies [103], hydra [104] and Tasmanian devils [105], indicates that animals with cancer may have a shorter lifespan, but many prioritize immediate reproductive efforts before succumbing to the illness. This adaptation, observed in host-parasite interactions, seems to also play a role in interactions with tumors, allowing for the maximization of offspring despite the presence of the disease.
Appendix C. Evolution of transmissible cancers
At present, there are fourteen documented instances of transmissible cancers in the wild. Two impact the Tasmanian devil [106] (Figure 3), one affects the Canidae [107], and eleven are identified in bivalves, some of which possess the ability to cross species barriers [108, 109]. The transmissibility of these cancer cells aligns their evolutionary dynamics more closely with emerging pathogens, fostering long-term co-evolution with their hosts. For example, after approximately forty years since its emergence, the transmissible cancer line associated with Devil Facial Tumor Disease (DFTD) in Tasmanian devils appears to transform into an obligatory parasite, engaging in genuine co-evolution with its host. Additionally, transmissible tumors have been mentioned in the freshwater cnidarian Hydra oligactis (Figure 4). Remarkably, these tumors and their distinct microbiome can be vertically transmitted during asexual reproduction (hydras generate buds that grow into distinct polyps). A surprising aspect of the external phenotype in certain tumorous hydras is the increased number of tentacles, which can escalate from 6–8 to around 20. A recent study [13], employing a tumor grafting protocol, demonstrated that the manipulation of the host phenotype is attributed to the tumor cells. The increased number of tentacles enables the hydra to capture more prey, facilitating increased budding and promoting tumor transmission. While tumor cells in general are known to manipulate their immediate environment, inducing processes like neoangiogenesis to nourish the tumor, this represents the first example of a tumor capable of influencing the external phenotype of its host, akin to how parasites manipulate behavior. This research supports the notion that transmissible cancers, due to their lifestyle remarkably similar to parasites, evolve under similar constraints as parasites sensu stricto, developing comparable adaptations through selection.
Tasmanian Devil (Sarcophilus harrisii), (A) healthy individual, (B) individual with a small tumor DFTD1 and (C) individual with a large tumor DFTD1 (Photo Credit, Frédéric Thomas).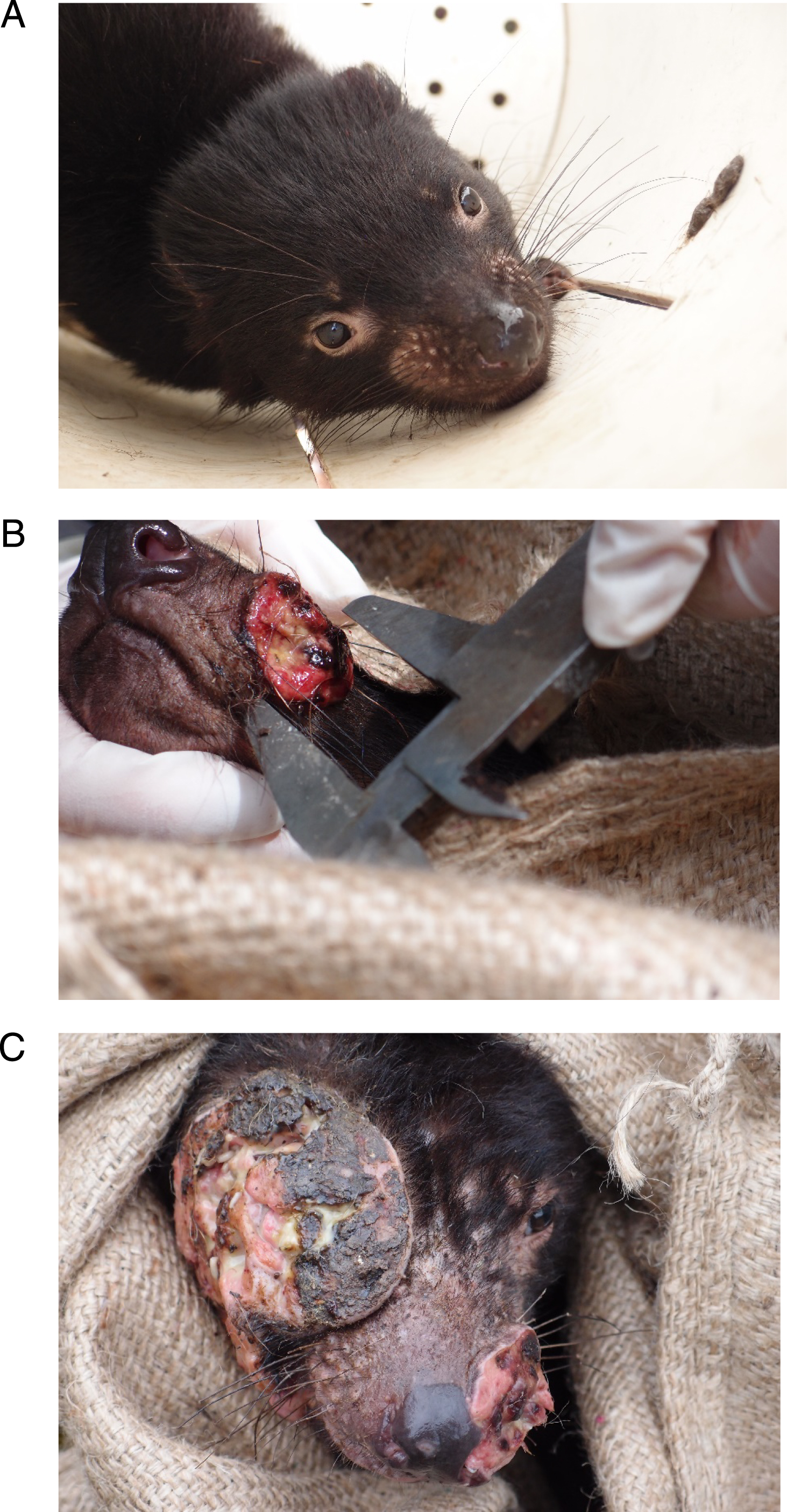
Hydra oligactis, healthy and tumoral polyps. In addition to the body being deformed due to tumors, we can notice the presence of additional tentacles, resulting from a manipulation of the host by tumoral cells [13].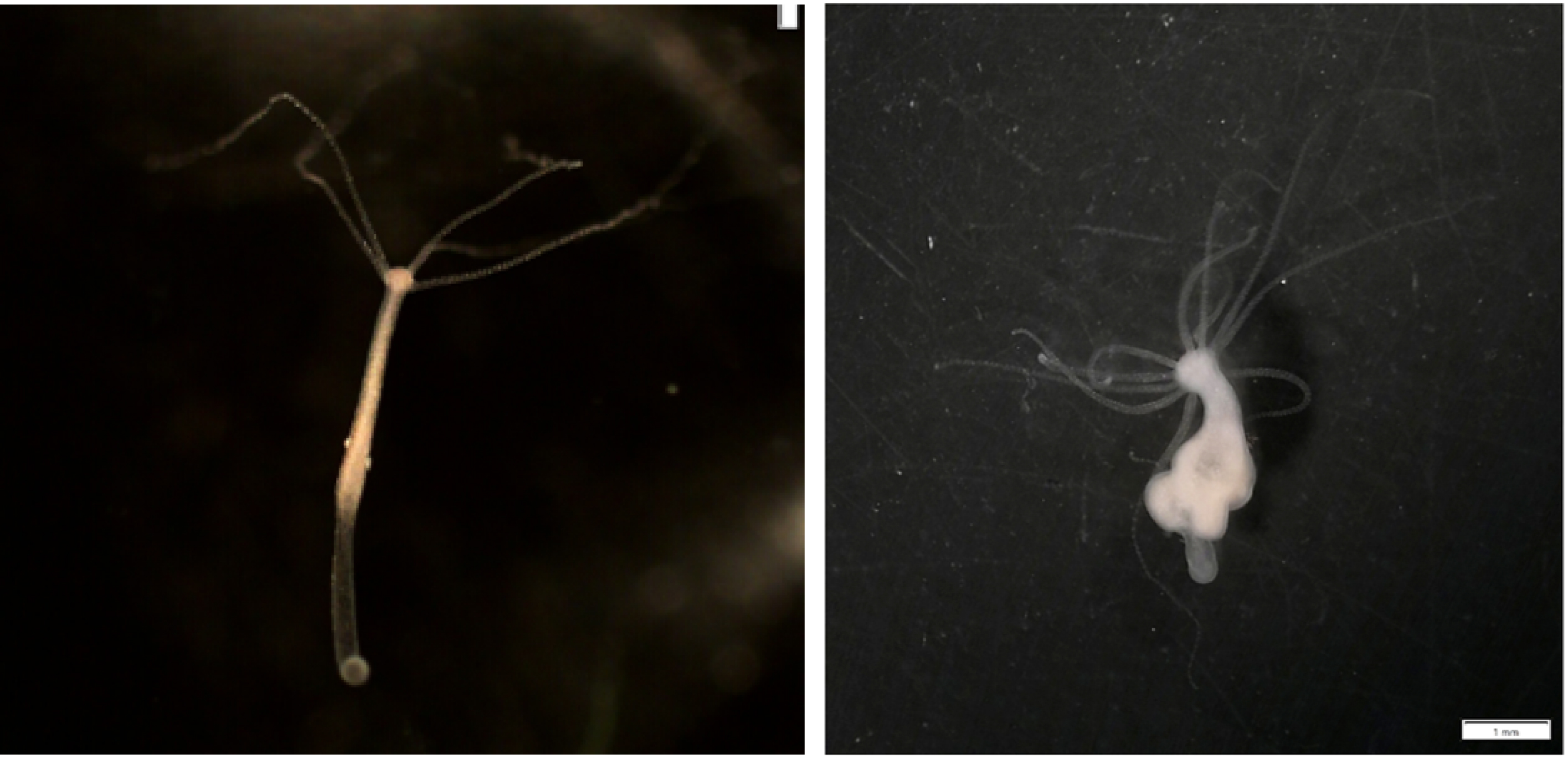
While the exact prevalence of transmissible cancers remains unknown, these unusual malignancies are likely to be rare in the wild. The underlying reasons for this rarity are only partly comprehended, and the “Perfect Storm hypothesis” proposes that transmissible cancers are infrequent due to the specific alignment of tumor and host traits necessary for their occurrence [111]. This explanation appears plausible, as transmissible cancers, akin to emerging pathogens, would require specific biotic and abiotic conditions not only for their emergence but also for their propagation to detectable levels [110] (Figure 5). Given the rarity of these conditions, transmissible cancers would infrequently spread and, most of the time, dissipate despite sporadic appearances. Therefore, further research is essential to identify the key factors that may either facilitate or impede the emergence and evolution of transmissible cancers. This inquiry gains significance as human activities progressively encroach upon wild habitats, causing alterations in ecosystems and their dynamics, potentially influencing the conditions conducive to the emergence and dissemination of transmissible cell lines, e.g., [112].
The emergence and spread of transmissible cancers hinge on a “Perfect Storm”, a precise set of conditions. Human activities have the potential to impact both of these crucial components (adapted from [110]).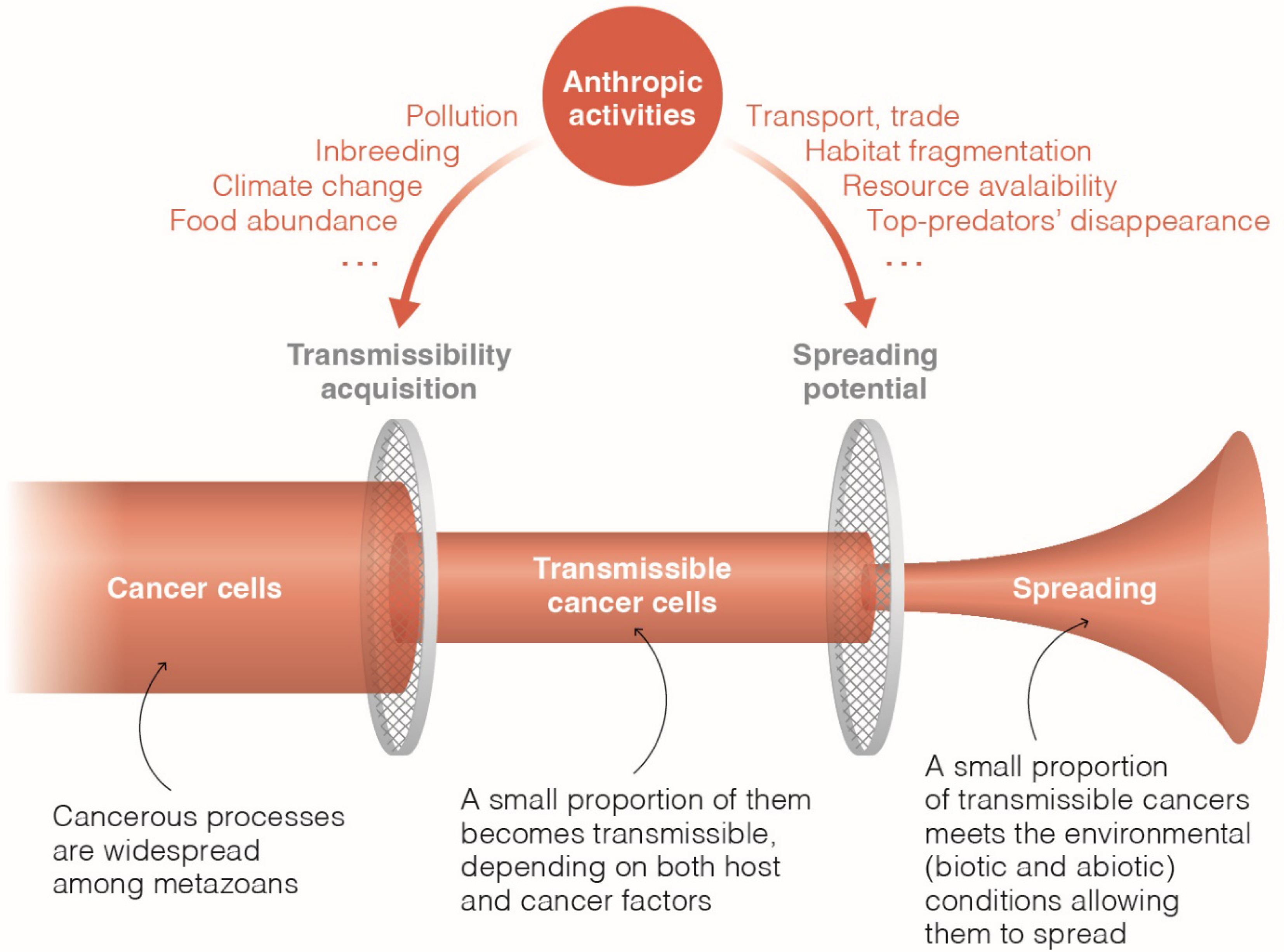
Appendix D. Ecological and evolutionary consequences of anticancer adaptations
Oncogenic processes are present across various branches of multicellular life. This phenomenon promotes the evolution of adaptations aimed at preventing or suppressing malignant progression and mitigating its impact on fitness. The significance of cancer cells in animal ecology has been largely overlooked by ecologists until recently, possibly due to a lack of consideration for the potential ecological and evolutionary repercussions of anticancer adaptations. However, evolution of anticancer adaptations has substantially constrained several facets of the evolutionary ecology of multicellular organisms at the cellular, individual, population, species, and ecosystem levels [15].
D.1. Cell level
Cancer defenses, while crucial for preventing tumors, can have unintended consequences and trade-offs that influence aging, regenerative capacity, and the prevalence of inherited mutations. These defenses are a double-edged sword, offering protection against cancer but at the same time, potentially accelerating the aging process. For instance, the P53 protein acts as a major cancer defense by interfering with normal cell activities, including cell cycle control, DNA repair, and the induction of cell death. This protein is a vital tumor suppressor, ensuring that cells with damaged DNA do not proliferate uncontrollably. However, its activation may have antagonistic pleiotropic effects, as it can impede the proliferation of normal cells, contributing to aging-related issues [62, 113]. When P53 is activated, it can lead to cellular senescence, a state in which cells permanently stop dividing. While this process prevents the formation and growth of tumors, it also means that the body’s ability to replenish cells is reduced. Over time, the accumulation of senescent cells can contribute to tissue degeneration and the functional decline associated with aging. Moreover, senescent cells secrete inflammatory cytokines, growth factors, and proteases, collectively known as the senescence-associated secretory phenotype, which can disrupt the tissue microenvironment and promote age-related pathologies. In addition to promoting senescence, the activation of P53 can also induce apoptosis, or programmed cell death, in cells with severe DNA damage. While apoptosis is a critical mechanism for eliminating potentially cancerous cells, excessive apoptosis can lead to a decrease in regenerative capacity. Tissues with a high turnover rate, such as the skin, blood, and gut, are particularly vulnerable to the loss of regenerative cells, resulting in impaired tissue repair and maintenance as we age. Furthermore, the trade-offs in cancer defenses can influence the prevalence of inherited mutations. High fidelity in DNA repair mechanisms reduces the risk of mutations that could lead to cancer. However, stringent DNA repair processes can also limit genetic diversity and adaptability, potentially impacting evolutionary fitness. The intricate balance between preventing cancer and allowing for cellular regeneration and repair underscores the complexity of the biological systems governing aging and disease. Thus, while mechanisms like the P53 pathway are indispensable for cancer prevention, they illustrate the inherent biological trade-offs that impact aging and senescence. Understanding these relationships is crucial for developing interventions that can mitigate the adverse effects of aging while maintaining effective cancer defenses [114, 115]. Additionally, cancer defenses that prevent neoplasm formation due to somatic mutations may paradoxically promote cancer. These defenses limit the detrimental effects of somatic mutations but also protect inherited mutations from purging by purifying selection, contributing to the higher frequency of oncogenic congenital mutations in populations [114].
D.2. Individual level
Individuals exhibit variations in their susceptibility to malignant cells and their ability to manage them, influenced by genetic and environmental factors [116]. This diversity extends to physiology, behavior, and life-history traits within species, affecting cancer risk and responses. For instance, individuals with high activity rates and metabolic rates tend to have lower cancer risk [117]. At the intraspecific level, different combinations of factors influence inter-individual performances, impacting traits like reproductive investment [16] and vulnerability to predators [36].
The evolution of prophylactic behaviors against cancer, such as habitat selection, has been explored, highlighting potential trade-offs [37]. Curative behaviors are considered more likely than preventive ones due to the association of malignancies with detectable internal cues. The effects of subclinical cancers on fitness in wild animals remain unknown, but potential trade-offs are expected, prioritizing cancer concerns over activities like reproduction. Self-medication in response to cancer is uncertain, and strategies to conserve energy or alter behaviors may emerge. Empirical studies, though limited, demonstrate altered reproductive schedules and social behaviors in individuals with early-stage tumors.
The influence of oncogenic processes on individual behavior in wildlife is underexplored, partly due to challenges in detecting cancer. Sexual selection is suggested as a promising avenue for exploration, considering potential trade-offs between energy allocation for cancer suppression and early-life reproductive activities (see for instance [118, 119]). Sexual selection may also favor oncogene alleles in certain cases, as observed in the fish genus Xiphophorus, where the oncogenic Xmrk allele persists in natural populations, suggesting benefits early in life that outweigh later costs through antagonistic pleiotropy [100].
D.3. Population level
Beyond individual-level plastic adjustments, the necessity for cancer defenses might shape the life-history of populations and species over the long term. Coevolving with oncogenic manifestations, individuals in certain populations may evolve a constant tumor-suppressive strategy, even in the absence of malignant cells.
One potential example of anticancer adaptations altering life-history traits involves Tasmanian devils heavily infected with DFTD, displaying a shift towards single breeding with individuals exhibiting precocious sexual maturity [105]. Exploring relationships between tumor development in wildlife and environmental contaminants, such as those from aluminum smelting facilities or organochlorine contaminants, reveals the intricate interplay of ecological factors, life-history characteristics, cancer defense mechanisms, and pollutant effects [120]. Local adaptations to cope with pollution are evident in various species, with fitness costs associated with these adaptations sometimes becoming apparent when individuals from polluted habitats are placed in non-polluted conditions. One of the most prominent illustrations of the swift and significant increase in the risk of mutagenic and potentially carcinogenic effects from modern human activities is the substantial release of radioactive isotopes into the air, soil, and water following the nuclear accidents in Chernobyl, Ukraine, in 1986, and Fukushima, Japan, in 2011 [121]. These incidents are notable not only for their profound impact but also for representing only the visible aspect of a broader issue. Human activities contribute to numerous mutagenic effects that may not be as conspicuous but should not be overlooked, given the implications of chronic exposure to low doses. Moreover, consensus is lacking on the long-term effects of chronic radiation exposure on wildlife in the Chernobyl Exclusion Zone and Fukushima area (see [122, 123]), highlighting the imperative for further research in these areas.
The theoretical concept of “evolved dependence” arises, suggesting that adapted individuals achieve higher fitness in the presence of oncogenic processes [120]. For instance, in Tasmanian devils with precocious sexual maturity, it might be expected that their fitness is lower than other individuals in the absence of the disease. This phenomenon, despite being poorly considered by ecologists, indicates local adaptations to cancer risk, which could be selected for in just a few generations in various organisms.
D.4. Species level
At the species level, it is commonly believed that the need to suppress somatic evolution should increase with larger body sizes and longer lifespans. However, as explained in Appendix B, “Peto’s Paradox” challenges this assumption, indicating no correlation between body size, longevity, and cancer rates across species. Recent studies reveal genomic mechanisms in large-bodied and long-lived species, such as elephants and whales, that enhance cancer suppression (see Appendix B).
While many studies explore adaptations in large and long-lived species to address size-associated cancer problems, few focus on the corollary consequences. Selection for anticancer mechanisms may act as a developmental and evolutionary constraint, potentially limiting the evolutionary trajectory of organisms. This constraint hypothesis is illustrated by the example of cervical vertebrae numbers in mammals [120].
The mismatch concept, often discussed in the context of recent changes in human evolution, also extends to natural environmental changes affecting species (see last section). Species may face natural mismatches between cancer risks and defenses due to evolving environmental conditions. Weak cancer defenses may still favor certain traits if fitness benefits outweigh the associated cancer costs or if compensatory adaptations evolve to limit these costs. Evolutionary constraints on anticancer defenses may lead to the evolution of compensatory fitness mechanisms, possibly explaining phenomena like menopause in humans and cetaceans [51]. The hypothesis posits that traits such as menopause, seen as a mechanism preventing the proliferation of malignant cells during pregnancies, are more readily selected than anticancer defenses. This implies that the selection for anticancer mechanisms presents an evolutionary constraint. The diversity of compensatory traits in the animal kingdom, when other cancer-associated traits cannot evolve due to insufficient cancer defenses, remains largely unexplored.
D.5. Ecosystem level
To comprehend the evolution of anticancer selection, it is essential to consider the complete ecological context, including the host’s community of organisms (parasites, microbiota) and the broader ecosystem (predators, competitors). This ecological approach allows a comprehensive assessment of selective pressures on anticancer selection. For instance, domesticated species, having often lived in oncogenic conditions but without the traditional constraints of the wild (competition, predation, parasitism), might experience a relaxation of these constraints [124]. This opens opportunities for the selection of novel anticancer defenses, as domesticated animals theoretically have more resources to invest in existing anticancer mechanisms. The maintenance of costly anticancer defenses is more likely in a domesticated setting compared to a natural ecosystem with various natural enemies. The impact of species interactions and the abiotic environment within ecosystems on the evolution of cancer avoidance remains largely unexplored but is crucial, considering species are exposed to these interactions over generations. Additionally, as most ecosystems are now polluted with mutagenic substances, the varying vulnerability of species to cancer initiation/progression, coupled with their ability to evolve cancer defenses, is likely altering biotic interaction equilibria. This, in turn, may lead to cascade effects, shaping population dynamics and communities—an emerging topic in ecology (see last section) (Figure 6).
Overview of the impact of cancer defense costs across various levels in the living world, spanning from cellular levels to individuals, populations, species, and ecosystems (adapted from [15]).