


 CC-BY 4.0
CC-BY 4.0
1. Reconstruire l’arbre du vivant : un objectif ambitieux initié par Charles Darwin
L’objectif ambitieux de reconstruire l’arbre du vivant, consiste, à terme, à représenter l’ensemble des 8 millions d’espèces vivantes sur un seul et même arbre évolutif [1] qui récapitulerait leurs relations de parenté – ou relations phylogénétiques. Dans cet arbre, les feuilles représentent les espèces actuelles, les nœuds leurs ancêtres communs. Établir un arbre des relations évolutives entre espèces est en premier lieu une façon de recenser et de classifier la diversité de la vie sur Terre. L’arbre du vivant est notamment précieux aux efforts de conservation de la biodiversité, car il documente de manière rigoureuse la diversité évolutive du vivant et permet ainsi de mieux comprendre comment celle-ci est affectée par les bouleversements environnementaux. En effet, la manière dont sont réparties les espèces menacées le long de l’arbre du vivant est une donnée importante pour prédire les branches les plus à risque de s’effondrer et induire les pertes les plus considérables en termes de diversité évolutive [2].
La première esquisse d’un arbre des espèces est attribuée à Jean-Baptiste de Lamarck, dès 1809 [3]. C’est seulement 50 années plus tard que Charles Darwin popularise définitivement le concept d’arbre du vivant, dans son ouvrage fondateur L’Origine des espèces [4]. Nous devons également à Ernst Haeckel de nombreux termes du domaine de la biologie évolutive ; inspiré par les écrits de Darwin, il propose notamment pour la première fois le terme de « phylogénie » [5]. Les premiers travaux phylogénétiques s’appuient sur les ressemblances anatomiques entre espèces pour déterminer leur proximité évolutive. Un siècle plus tard, la molécule d’ADN est reconnue comme un « document de l’histoire évolutive » : un levier puissant pour reconstruire les relations entre espèces [6]. En effet, il devient apparent que les mécanismes de modification et de transmission de l’ADN impliquent que la similarité de séquences d’ADN entre espèces est un indicateur de leur degré de parenté. S’ensuivent l’avènement de la phylogénie moléculaire, et une amélioration continue des modèles d’évolution des séquences, permettant d’inférer l’histoire de la vie sur Terre de manière toujours plus complète et plus exacte.
Malgré les progrès de la génomique et des techniques de séquençage de l’ADN, divers nœuds de la phylogénie des animaux continuent aujourd’hui de faire l’objet de débats considérables au sein de la communauté scientifique [7, 8]. Un des nœuds les plus controversés est probablement celui situé à la base de l’arbre des animaux : qui des éponges ou des cténophores ont divergé en premier du reste des espèces animales ? Si cette question anime les débats, c’est en partie parce que sa réponse pourra permettre d’éclairer l’origine évolutive des neurones et des muscles. Bien que morphologiquement assez distinctes [9], des cellules neuronales ainsi que des cellules musculaires sont présentes chez les cténophores et chez la plupart du reste des animaux, mais sont notablement absentes des éponges. Les différentes phylogénies proposées à la base de l’arbre des animaux impliquent différents scénarios évolutifs concernant l’origine des neurones et des muscles. Par exemple, l’hypothèse « divergence des cténophores en premier » implique soit une origine commune dans l’ancêtre des animaux avec une perte secondaire chez les éponges, soit une évolution indépendante dans chacun des deux groupes (les cténophores et le reste des animaux). Ainsi, la résolution de ce nœud fournira le cadre nécessaire pour évaluer ces scénarios évolutifs. Un second défi concerne la position de l’énigmatique ver marin Xenoturbella, surnommé « chaussette des mers » aux vues de sa morphologie simplifiée (pas d’œil, pas de système digestif, pas de cerveau). Ce dernier a été alternativement placé avec les mollusques, en groupe frère à l’ensemble des animaux bilatériens, ou encore proche du groupe des étoiles de mer. Enfin, de par leur impressionnante diversité, un travail considérable reste à accomplir afin de dresser un tableau complet de l’évolution du groupe que l’on nomme communément les poissons (poissons à nageoires rayonnées, actinoptérygiens) [10]. L’objectif de notre travail s’est focalisé sur la résolution d’un nœud récalcitrant situé à la base du plus grand clade parmi les poissons : celui de l’origine des poissons téléostéens.
2. Cinquante années de débats pour résoudre l’origine des poissons téléostéens
Le groupe des téléostéens regroupe plus de 96 % des espèces de poissons. Avec au total plus de 30 000 espèces recensées, il comprend autant d’espèces que les Tétrapodes (Amphibiens, Mammifères, Oiseaux et Reptiles) (Figure 1A). Les poissons téléostéens sont subdivisés en trois groupes : les Elopomorphes ou « anguilliformes » (tarpon, murène, anguille), les Osteoglossiformes ou « poissons à langue osseuse » (arowana, mormyre) et les Clupeocephala, qui regroupent la majorité des téléostéens (poisson zèbre, tétraodon, épinoche, morue, brochet…). On estime que le dernier ancêtre commun à ces trois groupes remonte à la période du Trias, il y a environ 250 millions d’années [11].
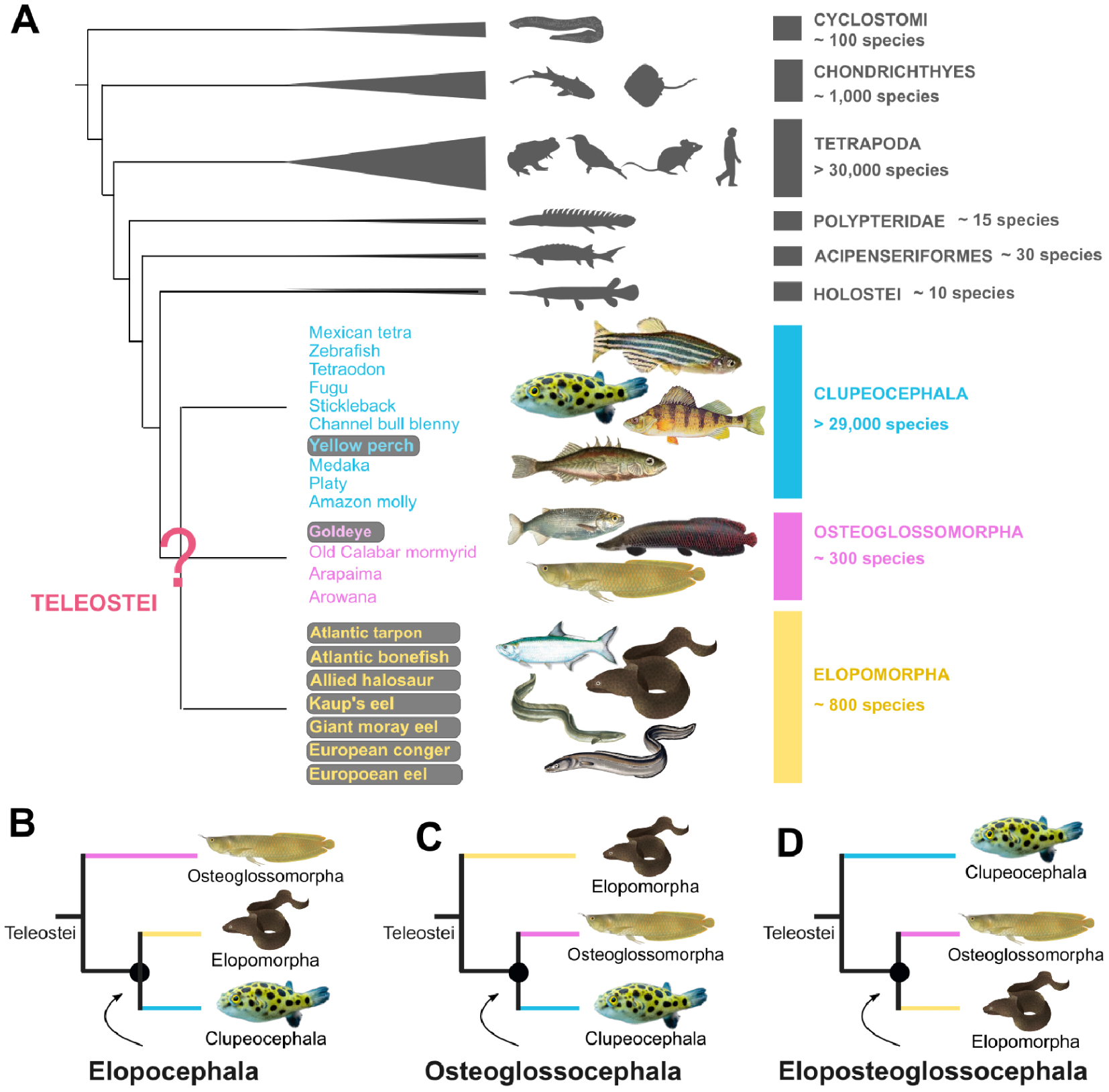
Position phylogénétique des poissons téléostéens au sein des Vertébrés. Les espèces listées pour illustrer chacun des trois groupes majeurs (Clupeocephala, Osteoglossomorpha, Elopomorpha) correspondent à celles considérées dans notre étude, dont celles pour lesquelles nous avons généré de nouveaux génomes (fond grisé). Les noms scientifiques complets des espèces incluses sont indiqués dans l’étude originale [12]. (B–D) Différentes topologies proposées à la base de l’arbre évolutif des poissons téléostéens : topologie Elopocephala [13, 14, 15], Osteoglossocephala [11, 16] et Eloposteoglossoceephala [17, 18, 19]. Figures adaptées à partir de [12].
La résolution des relations évolutives entre ces trois groupes représente un défi important, auquel se heurte la communauté depuis plus de 50 ans. Les premières études se basent sur des critères anatomiques et l’analyse de fossiles. Elles proposent le regroupement des Elopomorphes avec les Clupeocephala (Réf. [15] ; Figure 1B), puis celui des Osteoglossiformes avec les Clupeocephala (Réf. [13] ; Figure 1C). Cette question a été par la suite revisitée avec l’avènement des méthodes de phylogénies moléculaires qui, bien que considérant des quantités de données de plus en plus importantes, continuent de soutenir alternativement chacune des trois possibilités existantes pour regrouper ces espèces entre elles (Refs. [14, 17, 18, 11, 16, 19] ; Figure 1B,C,D).
Plusieurs hypothèses peuvent être mises en avant pour expliquer les incongruences constatées. Un premier argument propose que l’instabilité de la position des Osteoglossiformes et des Elopomorphes dans les phylogénies reconstruites serait due à leur large sous-représentation (en moyenne 14 fois moins d’espèces incluses par rapport aux Clupeocephala dans les études précédemment citées). D’autres considérations d’ordre méthodologiques pointent la diversité des méthodes employées, et la pertinence des choix techniques effectués pour modéliser l’évolution des séquences. Brièvement, deux familles principales d’approches sont généralement employées : (i) la méthode par « concaténation » qui considère l’ensemble des séquences d’ADN en un seul bloc pour reconstruire directement la phylogénie des espèces ; (ii) la méthode par « consensus », qui considère chaque gène séparément pour reconstruire un arbre pour chacun et en déduire la phylogénie. L’avantage de la méthode par concaténation est la quantité de données considérée qui maximise le signal phylogénétique, l’inconvénient est qu’elle repose sur l’hypothèse que l’ensemble des séquences d’ADN partagent la même histoire évolutive, une simplification rarement vérifiée dans la pratique. Par contraste, les arbres de gène de la méthode par consensus sont plus souvent affectés par des erreurs d’inférence dues à un plus faible pouvoir statistique, mais cette méthode permet de tenir compte d’histoires évolutives distinctes caractérisant différentes parties des génomes. Un phénomène nommé « tri de lignées incomplet » est le plus souvent responsable de ces histoires évolutives discordantes au sein d’un même génome. Le tri incomplet des lignées survient lorsque des différences génétiques préexistantes au sein d’une population ancestrale (différents allèles d’un gène par exemple) sont retenues différentiellement dans les espèces descendantes. Par exemple, si deux allèles A et B d’un même gène existaient dans la population ancestrale des poissons téléostéens et que chacun des trois grands groupes avait retenu soit l’allèle A soit l’allèle B de manière aléatoire, alors l’histoire évolutive de la séquence de ce gène ne suivra pas forcément la phylogénie des espèces. L’effet du tri de lignées incomplet est d’autant plus important lorsque plusieurs groupes d’espèces divergent les uns des autres dans un intervalle de temps court, comme cela a été le cas à l’origine des poissons téléostéens. Un phénomène biologique également susceptible de brouiller le signal phylogénétique est l’introgression : l’occurrence d’hybridation entre les populations anciennes des trois grands groupes aurait pu mener à des échanges d’ADN entre lignées, après leur séparation. Les méthodes consensus modélisent souvent le tri des lignées incomplet, mais rarement l’introgression.
Enfin, un des défis principaux de la reconstruction de phylogénies des poissons téléostéens est lié à leur histoire évolutive complexe : tous les téléostéens actuels descendent d’un ancêtre ayant connu un événement de duplication complète de son génome [20]. En conséquence, de nombreux gènes existent en deux copies dans les génomes de téléostéens, ce qui complexifie l’identification de gènes « marqueurs », c’est à dire comparables entre espèces et utilisables pour analyse phylogénétique. Plus précisément, le défi consiste à discerner les gènes orthologues (gènes qui descendent de la même copie du gène ancestral) des gènes paralogues (gènes qui descendent de copies dupliquées). L’événement de duplication qui sépare les gènes paralogues entre eux introduit une discordance entre l’histoire évolutive du gène et celui des espèces. Les séquences des gènes orthologues, quant à elles, reflètent directement l’histoire évolutive des espèces et sont donc de bons marqueurs pour reconstruire leur phylogénie.
Bien que les difficultés rencontrées lors de reconstructions phylogénétiques s’expliquent par une combinaison de plusieurs des facteurs précédemment cités, l’effet confondant des duplications complètes est formellement démontré [21] et a probablement largement contribué aux incongruences dans le cas des poissons téléostéens. Un parallèle peut être établi avec la diversification rapide des trois familles principales de Salmonidés dont les relations ont longtemps été ambiguës [22], en lien avec un événement de duplication complète de génome dans leur lignée ancestrale.
3. Une réanalyse à la lumière de nouvelles données génomiques
En vue de résoudre les relations phylogénétiques à l’origine des poissons téléostéens, nous avons mis en place de nouvelles analyses, conçues pour mitiger les limitations des études précédentes. Dans le cadre de notre étude, nous avons généré de nouvelles ressources génomiques, notamment pour le groupe des Elopomorphes pour lequel nous avons séquencé le génome de 7 espèces (Figure 1A). Au total, nous avons considéré 25 génomes (Figure 1A), en prenant soin de ne pas significativement surreprésenter un groupe par rapport aux autres : 7 Elopomorphes, 4 Osteoglossiformes, 10 Clupeocephala et 4 Vertébrés non-téléostéens.
La reconstruction d’une phylogénie moléculaire s’articule autour de 3 étapes principales : (i) l’identification de gènes marqueurs, présents et identifiables avec confiance dans tous les génomes considérés (gènes orthologues) ; (ii) l’alignement des séquences de ces gènes entre espèces, afin de mettre en évidence les positions qui ont changé au cours de l’évolution ; (iii) l’inférence d’un arbre phylogénétique sur la base des changements observés dans les séquences. Pour identifier des gènes marqueurs entre poissons téléostéens, tâche compliquée par leur événement ancien de duplication du génome, nous nous sommes appuyés sur des travaux que nous avions réalisés précédemment [23, 24]. Nous avons développé des méthodes spécifiquement adaptées aux particularités des génomes téléostéens, qui nous ont permis ici d’établir un jeu de gènes marqueurs à la fois plus complet et plus robuste que ceux des études précédentes. Cette méthode d’identification d’orthologues s’appuie sur une signature laissée dans les génomes par les événements de duplication complète. Initialement, l’ensemble des chromosomes sont dupliqués, puis, par la suite, les chromosomes dupliqués évoluent de manière indépendante et accumulent des pertes de gènes et réarrangements génomiques distincts, ce qui permet de les différencier et de les identifier entre espèces. Ainsi, nous identifions les gènes orthologues sur la base de leur conservation de séquence, mais aussi de la conservation de leur environnement génomique local, reflet de leur origine chromosomique commune [23, 24]. Grâce à cette approche, nous considérons un jeu de 955 gènes marqueurs, ce qui représente environ 5 % du répertoire génique complet, pour une taille totale des séquences géniques alignées de 2 328 657 nucléotides. En comparaison, les alignements analysés dans les études précédentes les plus compréhensives [18, 11, 19] comprenaient entre 500 000 et 1 000 000 nucléotides, soit environ 2 à 5 fois moins. Nous avons également appliqué une large gamme de méthodes différentes : nous avons reconstruit un total de 16 arbres phylogénétiques, en utilisant à la fois des approches de concaténation (inférence directe d’un arbre unique basé sur la totalité du jeu de gènes) et consensus (reconstruction d’un arbre par gène marqueur, réconciliés ensuite en une unique phylogénie). L’ensemble de ces analyses phylogénétiques ont toutes convergé vers la même topologie : la phylogénie Eloposteoglossocephala (Figure 1D). Bien que proposée par plusieurs études précédentes, il s’agit de la première fois que cette topologie est supportée indépendamment des méthodologies employées.
4. Résolution de l’origine des poissons téléostéens par le biais de méthodes innovantes
Ces nouvelles analyses phylogénétiques représentent un important pas en avant pour éclaircir les relations évolutives à l’origine des poissons téléostéens. Néanmoins, l’apport principal de notre travail réside dans la taille importante du jeu de données considéré (nombre de gènes), un atout mis en avant par chacun des travaux passés par rapport à leurs prédécesseurs. Dans l’objectif de rompre avec la dynamique des débats précédents, nous avons également tiré avantage de méthodes de phylogénies moléculaires innovantes, basées sur de nouveaux marqueurs.
Les méthodes de phylogénies moléculaires classiques se basent sur les changements observés dans les séquences d’ADN des gènes. Cependant, les séquences d’ADN ne sont pas les seules composantes des génomes à accumuler des modifications au cours du temps. La structure des génomes est également dynamique, impliquant, au fil des générations, une réorganisation de l’agencement des gènes le long des chromosomes. Ces modifications s’accumulent plus lentement que celles affectant les séquences, et offrent donc une perspective complémentaire pour étudier l’évolution des génomes. De plus, l’évolution de l’ordre des gènes est potentiellement moins affectée par l’introgression [25]. Bien que la démonstration de la pertinence d’examiner les changements structuraux des génomes pour retracer leur évolution est ancienne [26], son utilisation pour reconstruire les relations de parentés entre espèces est quant à elle très récente [27, 12, 28, 25].
Les mécanismes gouvernant l’évolution de l’agencement des gènes sur les chromosomes – l’évolution de la synténie – demeurent moins bien caractérisés que ceux entraînant des modifications de séquences. De ce fait, il n’existe pas de modèle probabiliste bien établi décrivant l’évolution de la synténie. En l’absence de modèle, nous avons appliqué des méthodes qui reposent sur l’estimation de distances évolutives entre paires de génomes, et visent à reconstruire l’arbre qui reflète au mieux ces distances. Ici, nous estimons les distances évolutives en quantifiant le degré de réorganisation de l’agencement des gènes entre deux génomes considérés (Figure 2A, 2B), et utilisons l’algorithme Neighbor-Joining [29] pour reconstruire un arbre à partir de cette matrice de distance (Figure 2C). L’algorithme Neighbor-Joining a été largement utilisé en phylogénie moléculaire classique, avant l’avènement des méthodes probabilistes. Nous appliquons également une méthode similaire (PhyChro, [27]), qui propose une amélioration par rapport à la méthode Neighbor-Joining, de façon à l’adapter aux marqueurs d’organisation des génomes. Notamment, dans l’algorithme PhyChro, le calcul de distance entre une paire de génomes examine également l’ensemble des autres génomes inclus dans l’analyse, afin de considérer spécifiquement les réarrangements génomiques informatifs à la reconstruction de l’arbre.
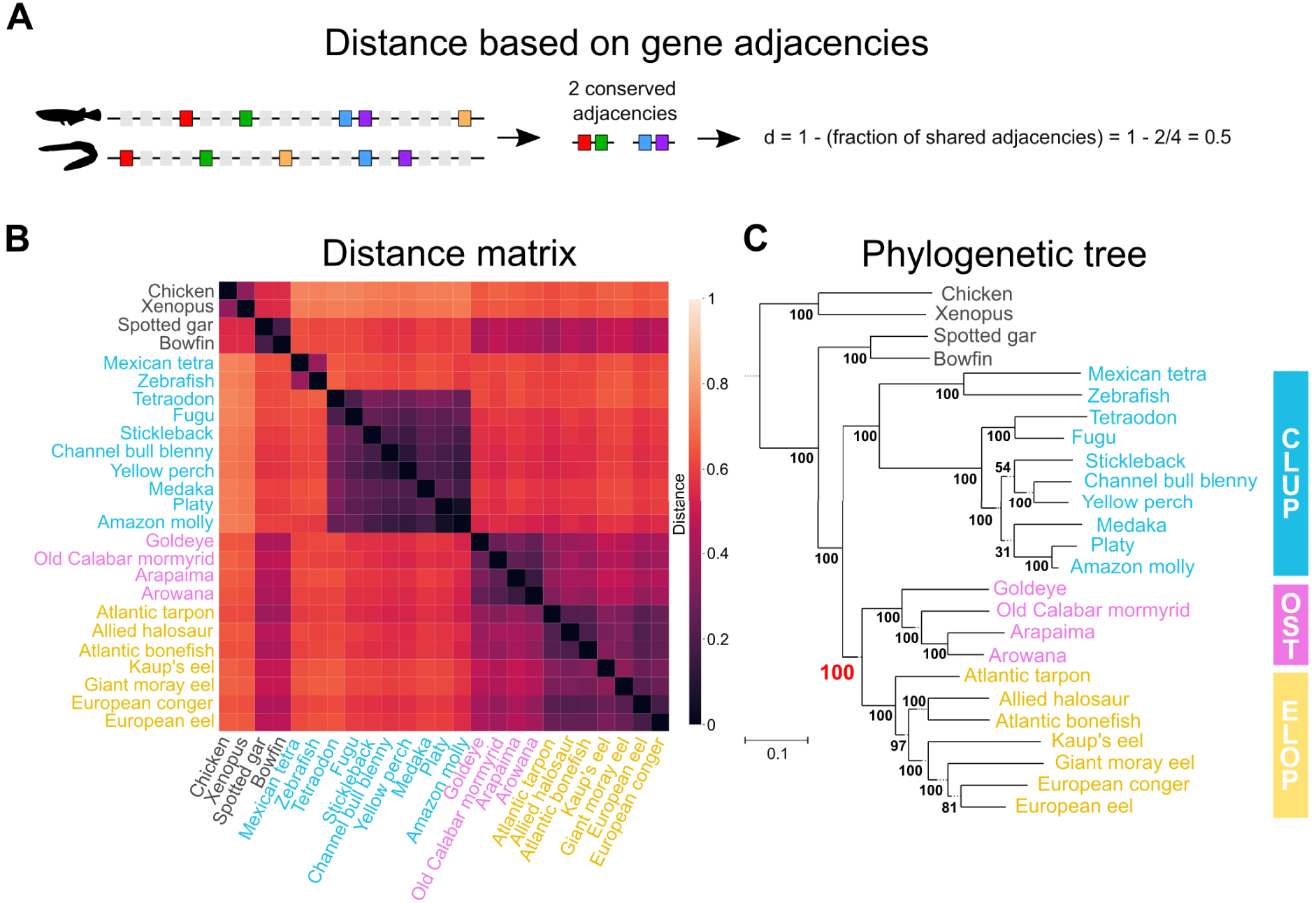
Phylogénie des poissons téléostéens basée sur l'évolution de l'ordre des gènes. (A) Définition d’une distance entre deux génomes basée sur la conservation des adjacences de gènes. Les gènes sont représentés par des rectangles le long d’un segment d’ADN, les gènes « marqueurs », c’est à dire identifiables dans tous les génomes considérés, sont indiqués en couleur. Une mesure de similarité est calculée sur la base de la proportion d’adjacences conservées entre gènes marqueurs dans les deux génomes comparés, puis transformée en distance. (B) Matrice de distances paire à paire obtenue pour les 25 génomes considérés et 3,041 gènes marqueurs, en utilisant la distance « d » définie en (A). La distance couverte bout à bout par les marqueurs considérés correspond en moyenne à 86 % du génome. (C) Arbre phylogénétique reconstruit par l’algorithme Neighbor-Joining [29]. à partir de la matrice de distance. Le groupe des Osteoglossomorpha est groupé avec celui des Elopomorpha (topologie Eloposteoglossocephala, Figure 1D), avec un fort support (100 % bootstrap).
Nous avons reconstruit un total de cinq phylogénies des poissons téléostéens basées sur l’organisation de leurs génomes, examinée à différentes échelles (notamment : adjacences conservées entre gènes marqueurs, agencements de blocs de gènes et organisation de chromosomes entiers). Dans chacune des cinq phylogénies ainsi reconstruites, nous retrouvons à nouveau la topologie Eloposteoglossocephala, qui regroupe les Elopomorphes avec les Osteoglossiformes (Figure 2C). Nous notons que ces différentes approches utilisent des jeux de gènes marqueurs identifiés par des stratégies différentes (voir [12] pour plus de précisions), ce résultat est donc robuste aux différents jeux de gènes considérés. En conclusion, à travers un large éventail d’analyses phylogénétiques complémentaires, nous avons pu résoudre les relations évolutives à l’origine des poissons téléostéens et ainsi démontrer que les anguilliformes sont plus proches des poissons à langue osseuse que du reste des poissons téléostéens.
5. Vers une utilisation plus large de la synténie pour résoudre l’arbre du vivant
Se servir de la structure des génomes comme levier pour reconstruire leurs relations évolutives nécessite un prérequis fondamental : les génomes considérés doivent être de qualité suffisante pour offrir une représentation fidèle et complète de la position des gènes le long des chromosomes. Ces ressources génomiques de haute qualité sont disponibles pour de nombreux groupes d’espèces, mais leur quantité est amenée à exploser dans les années à venir, grâce notamment aux nombreux projets de séquençage à grande échelle de la biodiversité (African BioGenome Project, ATLASea, Darwin Tree of Life, Earth Biogenome Project, European Reference Genome Atlas). Dans ce contexte, une utilisation plus systématique de la synténie pour reconstruire des phylogénies devient un objectif envisageable et prometteur, bien que représentant encore de nombreux défis méthodologiques [30]. En particulier, les nœuds profonds demeurent intrinsèquement difficiles à résoudre de par les périodes substantielles de temps écoulé, qui érodent le signal phylogénétique aussi bien dans les séquences d’ADN que dans la conservation de l’organisation des gènes. Pour que le signal biologique fourni par la synténie soit utilisable dans le cadre d’analyses phylogénétiques, l’ordre des gènes doit être variable entre les espèces étudiées sans pour autant être complètement dégradé. La définition d’indicateurs caractérisant les degrés de conservation et de dégradation limites représentera une avancée cruciale afin de délimiter le champ d’application optimal de ces nouvelles méthodes.
Une étude récente a examiné pour la première fois l’évolution de la synténie pour établir les relations évolutives à l’origine des animaux [28], un nœud daté à environ 650 millions d’années. Ces travaux ont mis en évidence des similarités entre l’organisation des génomes d’éponges et ceux du reste des animaux, suggérant une phylogénie dans laquelle les cténophores constituent le groupe-frère d’un clade réunissant les éponges et les autres animaux. Ce résultat surprenant, qui va à l’encontre des analyses les plus récentes de phylogénie moléculaire classiques [8, 31, 32], stimule les recherches afin de mieux comprendre les raisons méthodologiques et/ou biologiques sous-tendant ces incongruences. Les milliers de génomes de haute qualité disponibles dans un futur proche laissent entrevoir l’opportunité de mieux comprendre les mécanismes qui gouvernent l’évolution de l’organisation des génomes et ainsi réexaminer de nombreuses zones controversées de l’arbre du vivant.
Déclaration d’intérêts
Les auteurs ne travaillent pas, ne conseillent pas, ne possèdent pas de parts, ne reçoivent pas de fonds d’une organisation qui pourrait tirer profit de cet article, et n’ont déclaré aucune autre affiliation que leurs organismes de recherche.
Financement
Ce travail a été soutenu par l’Agence Nationale de la Recherche (ANR), dans le cadre du projet GenoFish 2016–2021 (grant No. ANR-16-CE12-003) et par le Conseil Européen de la Recherche, dans le cadre du programme de recherche European Union Horizon 2020 research and innovation (Grant Agreement No 817923, AQUA-FAANG). E.P. est actuellement soutenue par la Royal Society (Newton International Fellowship, NIF\R1\222 125).
Remerciements
Nous remercions tous nos co-auteurs de l’étude originale, pour leur contribution au travail que nous mettons en avant dans cet article de revue : Alexandra Louis, Jerome Montfort, Olivier Bouchez, Céline Roques, Carole Iampietro, Jerome Lluch, Adrien Castinel, Cécile Donnadieu, Thomas Desvignes, Christabel Floi Bucao, Elodie Jouanno, Ming Wen, Sahar Mejri, Ron Dirks, Hans Jansen, Christiaan Henkel, Wei-Jen Chen, Margot Zahm, Cédric Cabau, Christophe Klopp, Andrew W. Thompson, Marc Robinson-Rechavi, Ingo Braasch, Guillaume Lecointre, Julien Bobe and John H. Postlethwait.