


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Des défauts dans le processus complexe de la morphogenèse cardiaque sont à l’origine des malformations cardiaques congénitales qui affectent une naissance sur 100 [1]. Le cœur dérive du mésoderme, la plus centrale des trois couches germinales embryonnaires, et se forme dans la région antérieure de l’embryon précoce en tant que structure tubulaire avec des pôles veineux (la voie afférente) et artériel (la voie efférente). Les cellules progénitrices cardiaques situées dans le mésoderme splanchnique, en contact avec le futur endoderme de l’intestin antérieur, contribuent progressivement à la croissance du cœur. Ces cellules, appelées le second champ cardiaque (SHF), contribuent au myocarde du septum ventriculaire, du ventricule droit et de la voie efférente au pôle artériel, ainsi qu’au myocarde auriculaire, y compris à la septation auriculaire, au pôle veineux. Le déploiement des cellules du SHF coïncide avec la formation de la boucle cardiaque, un processus essentiel et conservé au cours duquel le tube cardiaque tourne vers la droite pour positionner correctement les futures régions du cœur définitif. Les étapes suivantes du développement du cœur impliquent la prolifération des cardiomyocytes entraînant la morphogenèse des chambres cardiaques, le développement des artères coronaires et du système de conduction cardiaque coordonnant les battements du cœur, et la septation cardiaque. Au cours de la septation, la formation des septa ventriculaires et auriculaires génère un cœur à quatre cavités, et la voie efférente embryonnaire est divisée pour former les voies efférentes des ventricules gauche et droit : l’aorte ascendante et le tronc pulmonaire. Le cœur embryonnaire, initialement tubulaire, acquiert ainsi des connexions indépendantes entre l’oreillette droite et le ventricule droit, et le ventricule gauche et l’aorte. À la naissance, la septation cardiaque est complétée avec la fermeture du canal artériel et du foramen interauriculaire, permettant la perfusion pulmonaire, et isolant ainsi les systèmes circulatoires pulmonaire et systémique.
L’allongement du tube cardiaque par l’ajout de cellules du SHF est nécessaire pour la septation cardiaque. Par conséquent, la perturbation du déploiement des cellules SHF chez les patients humains et les modèles animaux se traduit par un spectre de formes communes de cardiopathies congénitales [2, 3]. Ces anomalies incluent des malformations septales ventriculaires et auriculaires, ainsi que des défauts de la voie efférente, ou défauts conotroncaux, qui empêchent la séparation correcte de la circulation systémique et pulmonaire. Disséquer les programmes de développement qui contrôlent le déploiement normal des cellules progénitrices cardiaques est donc essentiel pour comprendre comment des défauts dans ce processus entraînent des maladies cardiaques, ainsi que pour diriger les cardiomyocytes dérivés de cellules souches vers différents destins régionaux pour les essais pharmacologiques et la réparation des tissus à la suite de lésions cardiaques. Nous passons ici en revue la contribution du SHF au cours du développement cardiaque et son implication dans les cardiopathies congénitales, en nous concentrant sur les rôles de deux facteurs de transcription à boîte T impliqués dans les anomalies cardiaques chez l’homme, TBX1 et TBX5.
2. Développement précoce du cœur et le deuxième champ cardiaque
Les cellules progénitrices cardiaques du mésoderme naissant émergent de la ligne primitive lors de la gastrulation et migrent vers la région antérieure latérale de l’embryon (Figure 1A). Deux vagues distinctes de cellules progénitrices ont été identifiées, activant indépendamment le facteur de transcription mésodermique précoce Mesp1 [4, 5, 6]. La première vague génère des cellules progénitrices du premier champ cardiaque (FHF), qui se différencient pour former des primordia cardiaques bilatéraux. Ceux-ci convergent sur la ligne médiane ventrale lorsque l’intestin antérieur se referme, donnant naissance au croissant cardiaque, et contribuant plus tard au myocarde ventriculaire gauche et au myocarde auriculaire.
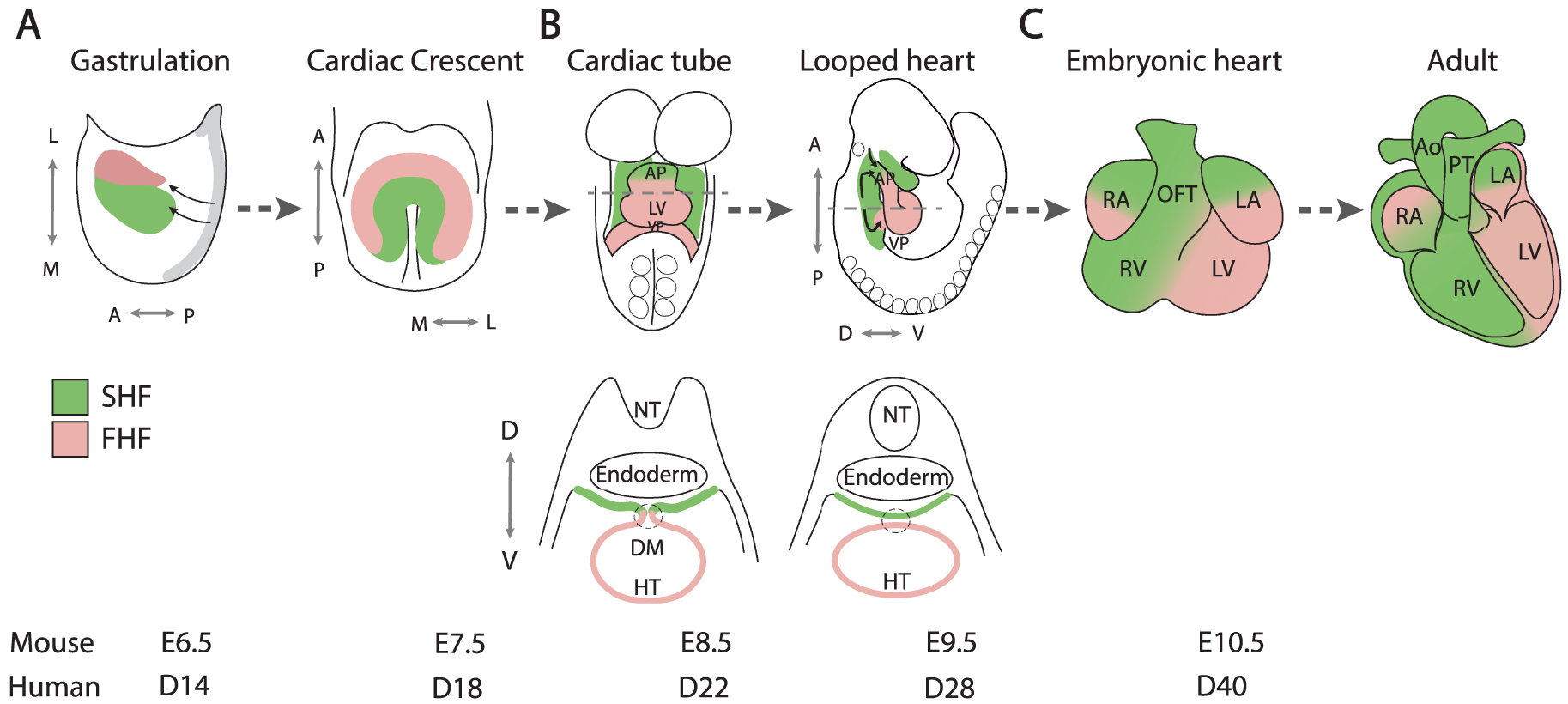
Développement cardiaque. Schéma montrant la contribution des cellules progénitrices du premier (rose) et du deuxième (vert) champ cardiaque au développement du cœur de la souris.(A) Morphogenèse du tube cardiaque ; (B) formation de la boucle et élongation du tube cardiaque ; et (C) croissance de la chambre et septation cardiaque. FHF, premier champ cardiaque ; SHF, second champ cardiaque ; L, latéral ; M, médial ; A, antérieur ; P, postérieur ; D, dorsal ; V, ventral ; AP, pôle artériel ; VP, pôle veineux ; HT, tube cardiaque ; RA, oreillette droite ; LA, oreillette gauche ; RV, ventricule droit ; LV, ventricule gauche ; NT, tube neural ; DM, mésocarde dorsal ; OFT, voie efférente ; Ao, aorte ; PT, tronc pulmonaire ; E : jour embryonnaire chez la souris, D : jour embryonnaire chez l’homme.
Le croissant cardiaque est transformé en un tube cardiaque, transitoirement linéaire, qui s’allonge ensuite et se courbe vers la droite, générant ainsi le cœur embryonnaire à la mi-gestation chez la souris, équivalent à la 5e semaine du développement humain (Figure 1B). L’allongement du tube cardiaque est contrôlé par l’ajout progressif de cellules du SHF, dérivées de la deuxième vague d’expression de Mesp1, situées dans le mésoderme pharyngé dorsomédial du tube cardiaque précoce, y compris dans la paroi dorsale de la cavité péricardique [5]. Le SHF a été identifié pour la première fois en 2001 grâce à l’analyse d’un transgène piège à activateur au locus Fgf10, exprimé dans le mésoderme pharyngé murin [7] et par des expériences de marquages cellulaires sur des embryons aviaires [8, 9]. Ces résultats s’appuyaient sur des études embryologiques pionnières montrant l’ajout de cellules extracardiaques aux pôles cardiaques [10, 11]. Contribuant initialement sur toute la longueur du primordium cardiaque, l’ajout du SHF se restreint aux pôles artériel et veineux après la rupture du mésocarde dorsal sépare le tube cardiaque de la paroi péricardique dorsale (Figure 1B). Des expériences de traçage génétique et de lignage rétrospectif ont montré que les deux champs cardiaques correspondent à deux lignages génétiques distincts définis par leurs contributions exclusives à la voie efférente et au ventricule gauche, et que les cellules progénitrices du SHF qui s’ajoutent aux pôles artériel et veineux sont clonalement apparentées [12, 13, 14]. Des expériences de marquage génétique et des analyses de lignage rétrospectives ont révélé l’ajout progressif de cellules progénitrices SHF au tube cardiaque en cours d’élongation, donnant naissance successivement au ventricule droit, à la voie efférente proximale et distale et au myocarde auriculaire, ainsi qu’aux cellules musculaires lisses du tronc pulmonaire (Figure 1C) [15, 16, 17, 18]. Des relations clonales ont été établies entre ces sous-populations de SHF et les muscles squelettiques branchiomériques de la tête et du cou, révélant ainsi l’existence de cellules progénitrices communes dans le mésoderme pharyngé [19, 20, 21, 22]. Cette hypothèse est renforcée par l’identification récente de cellules progénitrices multi-lignages qui contribuent au muscle squelettique et au myocarde, à l’aide d’une analyse de séquençage d’ARN sur cellule unique [23]. Le séquençage de l’ARN sur cellule unique est une approche révolutionnaire qui a permis d’obtenir des informations sans précédent sur l’hétérogénéité et les trajectoires de développement des populations de cellules progénitrices [24]. Cette technique a notamment permis l’identification d’une population de cellules du FHF appelée le champ juxtacardiaque, située à la limite latérale entre l’embryon et le mésoderme extraembryonnaire. Les cellules du champ juxtacardiaque contribuent à la croissance du primordium cardiaque précoce le long de sa marge ventrolatérale, contrairement aux contributions du champ SHF le long de la marge dorsomédiane [25, 26, 27]. Cette technologie a également révélé des différences dans l’expression des gènes entre les parties gauches et droites du SHF, ce qui est cohérent avec la découverte que le SHF est un moteur de la morphogenèse en boucle [28, 29]. Des défauts dans la formation de la boucle cardiaque, ou dans l’établissement préalable de la latéralité gauche droite embryonnaire, ont été impliqués dans un spectre de malformations cardiaques [29, 30]. Des expériences d’imagerie dynamique utilisant la microscopie fluorescente à deux photons et à feuillette de lumière ont commencé à confirmer les trajectoires empruntées par les cellules progénitrices cardiaques pendant la croissance du cœur précoce, révélant un délai temporel entre le déploiement des cellules du FHF et SHF, et des comportements cellulaires distincts dans le champ juxtacardiaque et le SHF pendant l’assemblage du tube cardiaque [6, 31]. En outre, les approches morphométriques quantitatives permettent une compréhension à haute résolution des événements complexes associés au développement précoce du cœur [32].
3. TBX1 et le développement du pôle artériel
Les cellules progénitrices qui s’ajoutent au pôle artériel du cœur, ou SHF antérieur, donnent naissance au myocarde du ventricule droit et de la voie efférente. Lorsque ces cellules convergent vers le pôle artériel, elles traversent une zone de transition dans laquelle l’expression des gènes des cellules progénitrices est régulée à la baisse et la différenciation myocardique est activée (Figure 2A) [33, 34]. Alors que l’absence de déploiement du SHF entraîne des phénotypes létaux précoces, des défauts plus légers dans ce processus génèrent une voie efférente trop courte pour permettre l’alignement normal entre le ventricule gauche et l’aorte ascendante lors de la septation cardiaque. Par conséquent, les défauts du SHF antérieur contribuent aux malformations conotruncales, qui représentent 30 % des malformations cardiaques congénitales, allant d’un désalignement ventriculo-artériel partiel, comme dans le cas de l’aorte chevauchante ou de la tétralogie de Fallot, à un ventricule droit à double issue (Figure 2B). Un défaut de croissance de l’aorte ascendante a également été impliqué dans la transposition des grandes artères [35].
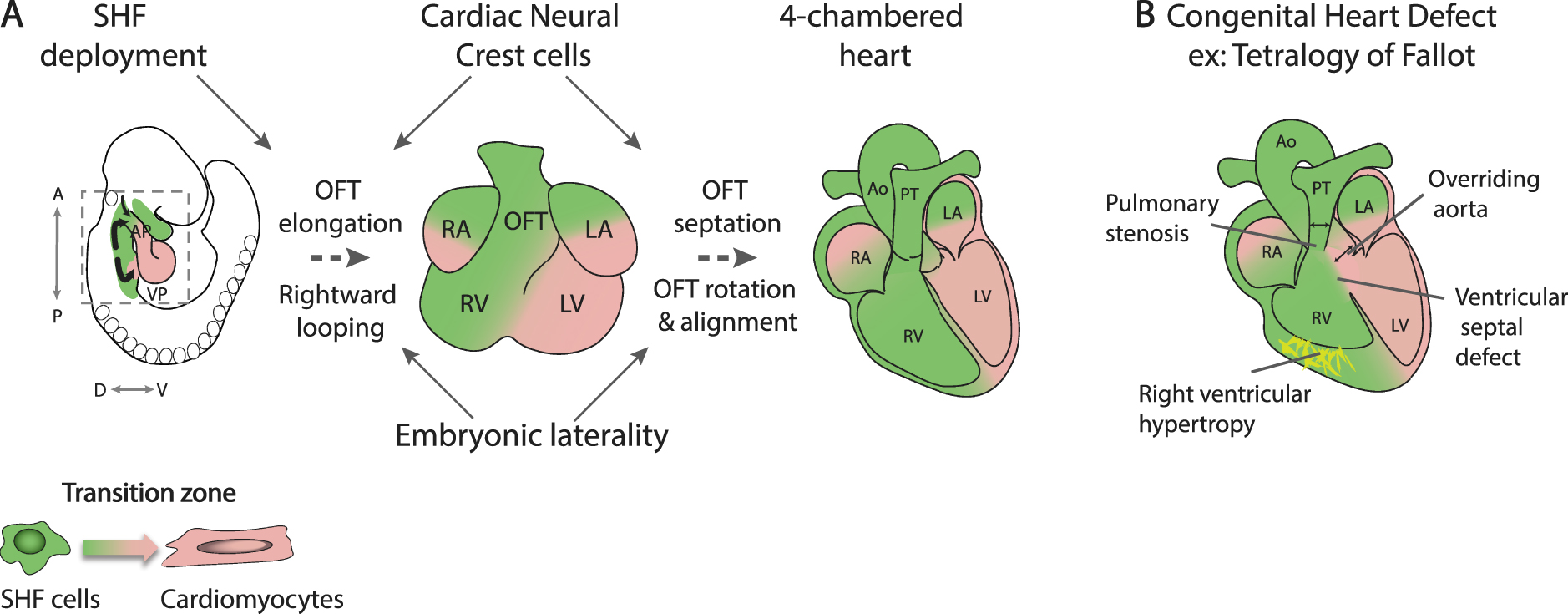
Développement du pôle artériel et septation de la voie efférente. (A) Alors que les cellules SHF contribuent à la paroi myocardique de la voie efférente au niveau de la zone de transition, l’ajout de SHF est régulé par les cellules de la crête neurale cardiaque nécessaire également pour la division de la voie efférente. La latéralité embryonnaire a un impact sur le looping et l’alignement ventriculo-artériel. (B) Exemple d’une malformation cardiaque congénitale conotroncale, la tétralogie de Fallot. SHF, Second champ cardiaque ; AP, pôle artériel ; VP, pôle veineux ; A, Antérieur ; P, Postérieur ; D, Dorsal ; V, ventral ; RA, oreillettes droites ; LA, oreillettes gauches ; RV, ventricule droit ; LV, ventricule gauche ; OFT, voie efférente ; Ao, aorte ; PT, tronc pulmonaire.
Les travaux menés par de nombreux groupes ont permis de définir les régulateurs transcriptionnels essentiels, les voies de signalisation intercellulaires et les caractéristiques épithéliales qui maintiennent l’état des cellules progénitrices du SHF et régulent la différenciation progressive aux pôles cardiaques [36, 37, 38, 39]. Outre l’expression de Fgf10, les cellules du SHF sont distinguées par l’expression des facteurs de transcription ISL1, TBX1 et d’un activateur transcriptionnel au locus Mef2c [7, 15, 16, 40]. Ces facteurs de transcription intègrent l’activité des voies de signalisation, y compris la signalisation autocrine et l’échange de signaux avec les cellules environnantes, en particulier l’endoderme pharyngé et le mésenchyme dérivé de la crête neurale. L’élongation du tube cardiaque est régulée par un équilibre entre les signaux FGF pro-prolifératifs et les signaux BMP pro-différenciation ; l’expression du FGF10 dans le SHF, par exemple, agit conjointement avec le FGF8 pour contrôler le développement du pôle artériel [41]. Lorsque les cellules SHF se différencient dans la zone de transition, l’expression de Fgf10 est régulée à la baisse par NKX2-5, qui bloque l’activation médiée par ISL1 d’un enhancer intronique de Fgf10 [42]. Il a été démontré que les cellules SHF de la paroi péricardique dorsale constituent un épithélium atypique, avec des jonctions serrées et d’adhérences apicales, et des filopodes basaux hautement dynamiques, plus typiques des cellules mésenchymateuses [43]. Les propriétés épithéliales des cellules du SHF apparaissent comme un point de régulation important contrôlant le déploiement du SHF. Ces propriétés sont régulées par des molécules d’adhésion cellulaire et des composants de la voie de polarité cellulaire planaire, tels que WNT5a et VANGL2, ainsi que TBX1, nécessaire à l’adhésion focale et à l’activité filopodale dans le SHF [33, 38, 43, 44]. En outre, l’analyse des jonctions apicales, de la forme des cellules, et de la distribution de l’actomyosine dans le SHF a suggéré des modèles biomécaniques régissant le déploiement du SHF, notamment, des mécanismes de poussée et de traction induits par la polarité cellulaire planaire et un circuit induit par la tension épithéliale favorisant la prolifération dans la région postérieure de la paroi péricardique dorsale [45, 46].
Les cellules de la crête neurale cardiaque, comme le SHF, se déplacent à travers la région pharyngée vers le pôle artériel cardiaque en développement, jouant un rôle important dans la division de la voie efférente embryonnaire en tronc pulmonaire et aorte ascendante [47]. L’ablation des cellules de la crête neurale entraîne donc la persistance d’un tronc artériel commun en raison d’un défaut de septation de la voie efférente ; l’ablation des cellules de la crête neurale cardiaque entraîne en plus un raccourcissement de la voie efférente en raison d’une réduction du déploiement du SHF (Figure 2A) [48, 49]. Cette observation chez les embryons aviaires a été l’une des premières à montrer l’importance de l’ajout de SHF pour la complétion de la boucle cardiaque et l’alignement de la voie efférente. Ces phénotypes reflètent l’échange de signaux entre ces deux types de cellules lorsqu’elles convergent vers le pôle artériel du cœur, orchestrant ainsi l’élongation normale de la voie efférente. Le facteur de transcription TBX1 a été identifié comme un régulateur critique du développement du SHF, favorisant la prolifération des cellules progénitrices et retardant la différenciation dans le SHF [40]. TBX1 est le principal gène impliqué dans le syndrome de délétion 22q11.2 ou syndrome de DiGeorge, caractérisé par des anomalies craniofaciales et cardiovasculaires, y compris une série de malformations conotroncales [50, 51]. Des expériences de séquençage de l’ARN sur cellule unique ont montré que Tbx1 contrôle les trajectoires myogéniques à partir de la population de cellules progénitrices multi-lignages vers des destins de muscles cardiaques et squelettiques [23]. Les embryons dans lesquels Tbx1 est absent présentent un tronc artériel commun associé à un développement défectueux des cellules de la crête neurale cardiaque ; en outre, les cellules qui donnent naissance au myocarde à la base du tronc pulmonaire, la sortie du ventricule droit, ne contribuent pas au pôle artériel du cœur mutant [40, 52, 53]. L’hypoplasie du myocarde sous-pulmonaire a été proposée comme un mécanisme en amont conduisant à la tétralogie de Fallot [54]. En effet, la tétralogie de Fallot est génétiquement hétérogène, mais la principale cause génétique connue est l’haploinsuffisance de TBX1, et le syndrome de délétion 22q11.2 représentant plus de 15 % des patients [50]. De plus, l’ablation du SHF dans les embryons aviaires conduit à un phénotype similaire à celui de la tétralogie de Fallot [55]. Des programmes génétiques distincts ont été définis dans le myocarde sous-pulmonaire et sous-aortique, reflétant des différences dans la distribution des cellules progénitrices dans le SHF ; par exemple, il a été démontré que Pparg et Dlk1 délimitent respectivement le myocarde sous-pulmonaire et sous-aortique [53, 56, 57]. Le futur myocarde sous-pulmonaire, initialement situé dans la paroi inférieure de la voie efférente, se positionne ventralement pendant la septation grâce à la rotation dans le sens inverse des aiguilles d’une montre de la paroi myocardique entraînée par l’ajout continu des cellules du SHF (Figure 2A) [58, 59].
Des études génétiques ont permis d’identifier des mutations associées aux coronaropathies sporadiques et familiales, notamment des mutations dans les gènes codant pour les facteurs de transcription T-box et les régulateurs de la chromatine [60]. Des exemples incluent TBX1 et CHD7, impliqué dans les défauts cardiovasculaires associés au syndrome CHARGE. CHD7 joue un rôle dans le remodelage de la chromatine dépendant de l’ATP, nécessaire aux contributions normales des lignées de la crête neurale et du SHF pendant le développement du cœur, y compris l’expression de Fgf10 dans le SHF par l’intermédiaire de l’enhancer intronique lié à ISL1 [61]. L’intégration d’études génétiques avec des données de séquençage d’ARN sur cellules uniques peut fournir des informations mécanistiques supplémentaires et a, par exemple, mis en évidence l’importance des cellules progénitrices de la voie efférente dans la tétralogie de Fallot [62]. Cependant, les défauts génétiques n’expliquent qu’un tiers des cas de cardiopathies congénitales à ce jour, ce qui suggère un effet important, mais mal compris, de l’exposition in utero à des facteurs environnementaux. En effet, des études récentes chez la souris ont identifié le SHF comme la cible d’une hypoxie gestationnelle de courte durée, d’une carence en fer maternelle et d’une exposition gestationnelle à l’alcool, entraînant dans chaque cas un déploiement réduit des cellules progénitrices du SHF et un raccourcissement de la voie efférente, ce qui se traduit par une série de malformations conotroncales [63, 64, 65].
4. TBX5 et septation cardiaque
DDes expériences de marquage par colorant et de traçage génétique des lignées ont montré que le myocarde sous-pulmonaire provient de cellules exprimant Hoxb1 dans la région latérale postérieure de la paroi péricardique dorsale, ce qui correspond à l’emplacement des cellules progénitrices multi-lignages [23, 53, 66]. La régulation à la baisse de l’expression de Hoxb1 est nécessaire au développement normal du pôle artériel et du ventricule droit [67]. Les cellules progénitrices cardiaques des lignées génétiques Isl1, Hoxb1, Tbx1, Wnt5a et Mef2c contribuent non seulement au pôle artériel mais également au pôle veineux du cœur, apportant des contributions essentielles aux structures septales auriculaires, dont le septum auriculaire primaire et la protrusion mésenchymateuse dorsale qui relie le septum auriculaire aux coussins auriculo-ventriculaires (Figure 3A) [53, 67, 68, 69, 70]. Des défauts dans le développement de la protrusion mésenchymateuse dorsale entraînent des malformations septales auriculaires de type primum, une classe de malformations septales auriculaires [71].
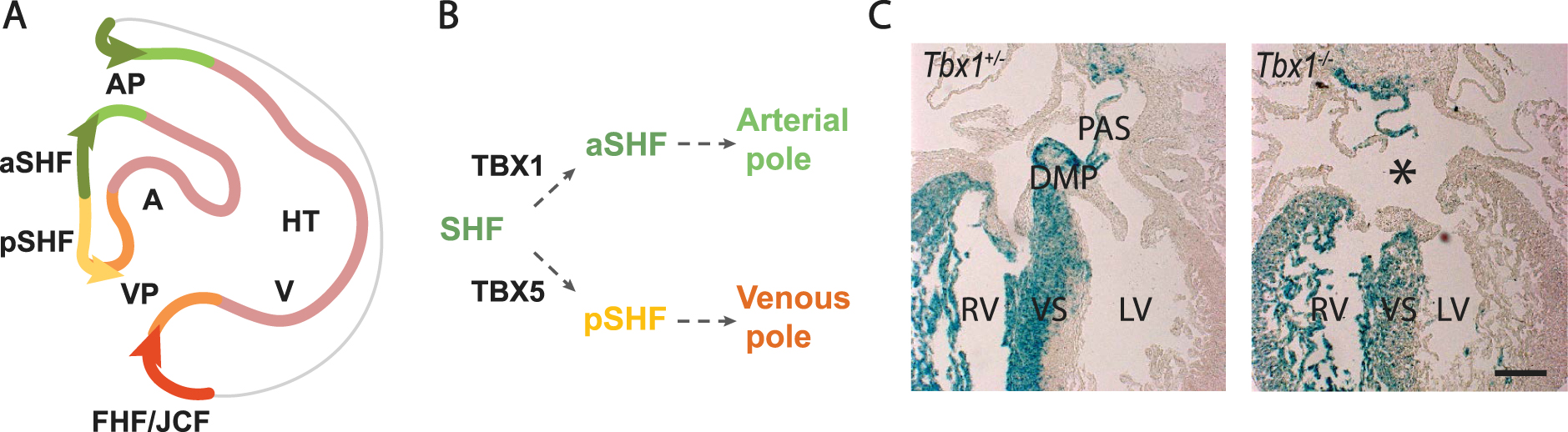
Développement du pôle veineux et septation auriculaire. (A) Schéma de la région encadrée dans la Figure 2A, montrant la ségrégation des cellules progénitrices en domaines du antérieur (aSHF) et postérieur (pSHF), contribuant aux pôles artériels et veineux. Notez la contribution des cellules du premier champ cardiaque (FHF) du champ juxtacardiaque (JCF) au pôle veineux. (B) Diagramme montrant que l’expression de Tbx1 est maintenue dans le SHF antérieur, tandis que Tbx5 est activée dans le SHF postérieur. (C) À gauche : la lignée génétique du SHF (bleu) marquée par un gène rapporteur conditionnel β-galactosidase activé par la recombinase Cre dans le SHF, montrant le marquage dans le ventricule droit (RV), le septum interventriculaire (IVS) et les structures septales auriculaires : le septum auriculaire primaire (PAS) et la protubérance mésenchymateuse dorsale (DMP). À droite : le DMP ne se forme pas dans les cœurs Tbx1 mutants, ce qui entraîne une anomalie du septum interauriculaire. AP, pôle artériel ; VP, pôle veineux ; A, oreillette ; V, ventricule ; RV, ventricule droit ; LV, ventricule gauche. Barre d’échelle en C : 100 μm.
Contrairement au SHF antérieur, qui conserve le programme génétique précoce du SHF, les cellules du SHF postérieur activent Tbx5, un régulateur clé du programme de différenciation du FHF et du pôle veineux, sous l’influence de la voie de signalisation de l’acide rétinoïque [72, 73]. Tbx5 est régulé à la hausse dans le mésoderme pharyngé postérieur exprimant TBX1 lorsque le mésocarde dorsal se décompose et que les cellules progénitrices ségrégent vers des pôles alternatifs (Figure 3B). La co-expression transitoire de TBX1 et TBX5 est suivie d’une régulation négative du programme du SHF antérieur et l’établissement d’une frontière transcriptionnelle au sein de la paroi péricardique dorsale. Cette frontière sépare les progéniteurs du pôle artériel positifs pour TBX1 dans le SHF antérieur des progéniteurs du pôle veineux positifs pour TBX5 dans le SHF postérieur [73]. Les mutations de TBX5 entraînent le syndrome de Holt-Oram, caractérisé par un défaut de septation auriculaire et des anomalies des membres antérieurs [74]. Il a été démontré que TBX5 active l’expression d’Aldh1a2, qui code une enzyme de biosynthèse de l’acide rétinoïque, ce qui suggère un mécanisme de rétroaction entre TBX5 et la signalisation de l’acide rétinoïque [75]. La frontière entre les domaines antérieur et postérieur du SHF est perturbée en l’absence de Tbx1, ce qui entraîne l’échec du développement de la protrusion mésenchymateuse dorsale et des malformations de la septation interauriculaire dans 30 % des embryons dans lesquels Tbx1 est absent, ce qui pourrait expliquer l’occurrence des malformations du septum interauriculaire chez les patients avec le syndrome de délétion 22q11.2 (Figure 3C) [53]. Des anomalies similaires du septum auriculaire sont observées dans les embryons de souris mutants conditionnels dépourvus de Tbx5 dans le SHF [76]. Le SHF postérieur partage des contributions de lignage avec le mésenchyme pulmonaire et l’échange de signaux entre le SHF et l’endoderme pulmonaire adjacent joue un rôle important dans le développement du pôle veineux [75, 77]. TBX5 active l’expression du ligand Wnt dans la partie postérieure du SHF qui signale à l’endoderme pulmonaire, contrôlant à son tour le moment de la différenciation dans la partie postérieure du SHF par le biais de la voie de signalisation Hedgehog [78, 79].
Des cellules exprimant à la fois Tbx5 et le programme SHF ont également été identifiées dans la région du septum interventriculaire à l’aide d’une approche de rapporteur de lignage intersectionnelle [4]. L’inactivation conditionnelle avec la recombinase Cre entraînée par l’enhancer Mef2c a montré que Tbx5 est nécessaire dans le SHF pour la formation du septum ventriculaire musculaire ainsi que pour la septation auriculaire [76, 80]. De plus, les facteurs de transcription MEF2C et TBX5 corégulent les gènes cibles nécessaires au développement du septum ventriculaire [81]. Ainsi, les structures septales auriculaires et ventriculaires se forment à des sites où l’expression de Tbx5 et le programme SHF se chevauchent transitoirement, ce qui suggère que cela définit un programme frontalier dans le cœur embryonnaire, nécessaire pour initier la morphogenèse septale. Les défauts de septation auriculaires et ventriculaires représentent ensemble plus de 50 % des malformations cardiaques [82]. Une dissection plus poussée du programme à l’interface des champs cardiaques sera essentielle pour améliorer notre compréhension des origines des malformations septales.
Conclusions et perspectives
Depuis la découverte du SHF, de nombreuses observations ont révélé l’importance de cette population de cellules progénitrices spatiotemporellement hétérogènes dans l’étiologie des maladies cardiaques affectant le développement des pôles du cœur et des septa cardiaques. Alors que les défauts graves d’élongation du tube cardiaque entraînent une létalité embryonnaire précoce, des perturbations plus légères de l’ajout de cellules aux pôles cardiaques entraînent des défauts d’alignement et l’absence de séparation complète de la circulation systémique et pulmonaire à la naissance, ce qui contribue à un spectre de formes communes de malformations cardiaques rencontrées chez les patients humains. Des progrès technologiques, tels que la transcriptomique sur cellule unique et les approches morphométriques quantitatives, ainsi que l’imagerie dynamique et l’établissement d’une variété de modèles d’organoïdes cardiaques dérivés de cellules souches, fournissent de nouvelles informations sur la régulation du développement du SHF [31, 83]. Les objectifs actuels comprennent l’élucidation des réseaux de régulation contrôlant des sous-populations distinctes du SHF, l’identification des voies en aval affectant la morphogenèse régionale du cœur et la septation cardiaque, et la dissection des mécanismes pathologiques par lesquels les facteurs génétiques et environnementaux interagissent dans le SHF pour provoquer les défauts morphologiques.
Déclaration d' intérêt
Les auteurs ne travaillent pas, ne conseillent pas, ne possèdent pas d'actions ou ne reçoivent pas de fonds de toute organisation qui pourrait bénéficier de cet article, et n'ont déclaré aucune affiliation autre que leurs organismes de recherche.
Financement
Les projets de l’équipe de RK sont soutenus par l’Agence Nationale pour la Recherche (projets PP-heart ANR-20-CE13-0029-01 et Heartbound ANR-22-CE13) et l’AFM-Téléthon. CG reconnaît le soutien du programme du gouvernement français géré par l’Agence Nationale de la Recherche (ANR-16-CONV-0001) et l’Initiative d’Excellence d’Aix-Marseille Université A*MIDEX (Turing Centre for Living Systems) et la Fondation pour la Recherche Médicale. RK est chercheur à l'Inserm.