

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Le cancer demeure l’une des principales causes de mortalité dans le monde, englobant diverses pathologies caractérisées par l’altération de processus contrôlant l’homéostasie cellulaire et tissulaire tels que la division cellulaire ou la croissance [1]. Son initiation est généralement localisée dans un organe ou un tissu spécifique, mais peut ensuite se propager à d’autres parties du corps. Connu depuis les civilisations anciennes, égyptienne, grecque, romaine, ainsi que dans la médecine traditionnelle chinoise, le cancer a été observé sous un nouveau jour avec l’avènement de la microscopie au 19e siècle, permettant notamment la détection des métastases. Durant ces premières phases de recherche sur la tumorigenèse, plusieurs hypothèses furent formulées [2]. C’est à Theodor Boveri qu’est attribuée la première formulation de la théorie majoritairement acceptée sur l’origine des cancers, la « théorie des mutations somatiques » [3]. Cette théorie postule que le cancer débute par un changement chromosomique dans une cellule, favorisant sa transformation tumorale et se transmettant à ses cellules filles.
Outre les mutations de l’ADN, les altérations épigénétiques jouent également un rôle crucial dans le cancer. L’épigénétique est la discipline qui étudie les mécanismes et les molécules permettant l’héritage de différents profils d’expression génique à partir de la même séquence d’ADN [4]. Ces mécanismes sont essentiels non seulement au cours du développement et à l’âge adulte, mais aussi dans le processus de vieillissement et dans la majorité des pathologies humaines, y compris le cancer [5]. Bien que l’épigénétique soit souvent considérée d’intérêt thérapeutique en raison de son rôle dans la progression des tumeurs et les métastases [6, 7], des données récentes, que seront discutées dans cet article, suggèrent que des altérations épigénétiques peuvent également initier le processus de tumorigenèse. Ces découvertes invitent à reconsidérer le rôle de la séquence d’ADN dans la progression tumorale et à repenser nos stratégies de prévention et de traitement des cancers.
2. La théorie des mutations somatiques et son influence sur la cancérologie moderne
Dans sa première formulation en 1914 [3], Theodor Boveri postulait que le cancer pouvait résulter de défauts chromosomiques, en particulier des problèmes de ségrégation des chromosomes lors des divisions cellulaires. Cette hypothèse a reçu une première confirmation avec la découverte de chromosomes aberrants dans des leucémies [8, 9] et, plus tard, avec l’identification des premiers oncogènes par plusieurs laboratoires [10]. Ces découvertes ont été intégrées dans la « théorie des mutations somatiques » (somatic mutation theory ou SMT) qui soutient que les cancers naissent de mutations génétiques. Il est néanmoins important de souligner que Boveri ne se focalisait pas uniquement sur l’ADN. Les chromosomes sont également constitués de protéines et d’ARNs qui jouent un rôle essentiel dans la régulation de l’expression génique et le maintien de l’intégrité chromosomique. A partir des années 1980, la recherche intense sur les oncogènes et les gènes suppresseurs des tumeurs a renforcé l’hypothèse que tous les cancers seraient causés par des mutations, et que la compréhension des cancers pouvait être avancée en dressant un catalogue exhaustif des oncogènes et des suppresseurs de tumeurs.
Dans un article publié en 1976 [11], Peter Nowell formule l’hypothèse que le développement des cancers se déroulerait en plusieurs étapes. Une première cellule (ou un petit groupe de cellules) subirait une modification initiale qui la rendrait néoplasique, lui conférant un avantage prolifératif. Par la suite, d’autres modifications, probablement dues à des mutations, déclencheraient un processus de sélection clonale, menant finalement au développement de tumeurs malignes. Cette publication est une pierre angulaire en oncogenèse car elle formule l’existence de cellules initiatrices de tumeurs et de leur évolution clonale. Suite à la découverte des oncogènes et des gènes suppresseurs de tumeurs, une fusion s’est produite entre l’hypothèse de l’initiation clonale de la tumorigenèse et la théorie des mutations somatiques, qui apparaîtraient dans la ou les cellules initiatrices des cancers (Figure 1). Les hypothèses alternatives suggérant une origine non-génétique du cancer, impliquant un dérèglement de la régulation génique, furent écartées. La notion que les tumeurs contiennent des clones de cellules ayant prédominé dans une « compétition évolutive » a ensuite renforcé la conviction de la communauté scientifique dans l’idée que le séquençage massif des génomes tumoraux permettrait d’identifier l’ensemble des gènes responsables du cancer.
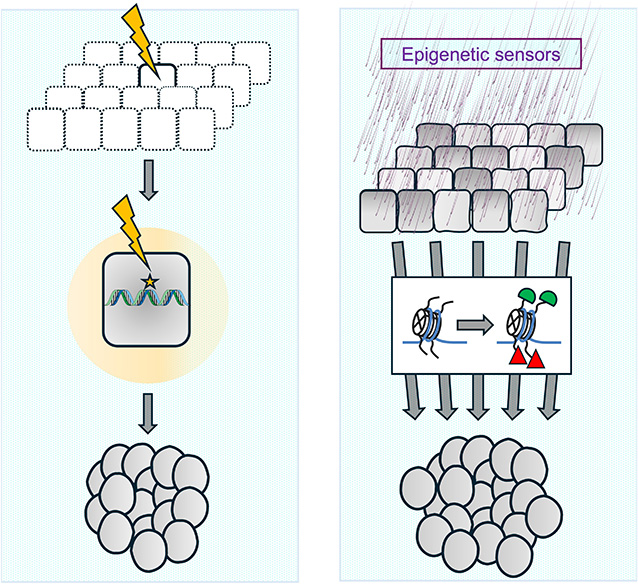
Origine mutationnelle ou épigénétique des cancers. A gauche, représentation de l’hypothèse dominante sur l’origine des cancers, qui s’appuie sur la théorie des mutations somatiques. Selon cette théorie, des mutations stochastiques, éventuellement provoquées par une exposition à des agents mutagènes (représentés en haut par une flèche jaune), pourraient parfois induire des oncogènes ou inactiver un gène suppresseur de tumeurs, générant la cellule initiatrice des tumeurs ou « cancer stem cell » (au centre). Suite à d’autres événements mutagènes et à une sélection des clones cellulaires, un cancer agressif se développe in fine (en bas). A droite, représentation de l’hypothèse de l’origine épigénétique des cancers. L’exposition à des agents carcinogènes non mutagènes, des changements nutritionnels et métaboliques ou d’autres sources de stress (en haut) pourraient affecter un ensemble de cellules qui changent l’état chromatinien d’une partie de leurs gènes (au centre, les modifications épigénétiques modifiées sont représentées en vert et en rouge). Ceci induit des changements d’expression génique qui, s’ils sont capables de s’auto-maintenir, pourraient initier un processus de tumorigenèse se maintenant même après la fin du stimulus ayant produit la première altération épigénétique. Masquer
Origine mutationnelle ou épigénétique des cancers. A gauche, représentation de l’hypothèse dominante sur l’origine des cancers, qui s’appuie sur la théorie des mutations somatiques. Selon cette théorie, des mutations stochastiques, éventuellement provoquées par une exposition à des agents mutagènes (représentés en ... Lire la suite
Ainsi, de la même manière que l’on oppose souvent le néodarwinisme au darwinisme dans le domaine de l’évolution (Encart 1), la SMT pourrait être envisagée comme une vision « néoboverienne » où seules les mutations d’ADN sont responsables du cancer, réfutant le rôle causal d’autres altérations telles que la composition ou la structure des chromosomes.
Encart 1
Les concepts Darwiniens font référence à la théorie de l’évolution des espèces par sélection naturelle, telle qu’élaborée par Charles Darwin dans L’Origine des espèces [12].Cette théorie repose sur l’observation des variations phénotypiques entre espèces, notamment des séries d’espèces similaires, suggérant que des traits phénotypiques se modifient au fil de générations. Dans la théorie Darwinienne, les caractères sont transmis par un mécanisme appelé pangenèse, où l’ensemble de l’organisme participerait à l’hérédité, notamment par le bourgeonnement de gemmules à partir de ses cellules, en particulier dans les organes génitaux.
Les concepts néo-Darwiniens découlent de la théorie synthétique de l’évolution. Cette version moderne de la théorie de Darwin intègre les découvertes génétiques postérieures, notamment la théorie mendélienne de l’hérédité ainsi que la génétique des populations, comme fondement de l’évolution [13, 14].Dans cette synthèse moderne de la théorie de l’évolution, la source des variations entre individus d’une même espèce réside dans les différentes informations génétiques transmises dans les gamètes. En général, on considère que ces informations sont contenues dans la séquence d’ADN et que les variations génomiques sont augmentées par des mécanismes aléatoires dus aux mutations et à la recombinaison méiotique. Les mécanismes de sélection dépendent de l’aptitude à survivre et à se reproduire de chaque individu au sein de populations de chaque espèce. Cette aptitude dépend de celles d’autres individus, de la taille des populations, ainsi que des conditions environnementales. L’épigénétique montre que des informations autres que celles présentes dans la séquence d’ADN peuvent contribuer aux caractéristiques phénotypiques. Sa contribution à l’évolution est aujourd’hui un sujet de discussion au sein de la communauté scientifique.
Avec l’avènement des techniques de séquençage à haut débit, de vastes projets de séquençage de cohortes de différents types de cancer ont été lancés. Ces projets ont rapidement permis l’identification de gènes fréquemment mutés, amplifiés ou délétés dans les échantillons de cancers [15, 16]. Parallèlement, des études fonctionnelles ont montré que ces mêmes altérations génétiques peuvent provoquer la formation de tumeurs chez la souris [17]. Ces preuves expérimentales, soutenues par des données épidémiologiques, confirment que les mutations des séquences d’ADN jouent un rôle majeur. Elles sont régulièrement utilisées comme biomarqueurs pour le diagnostic et le pronostic en pratique clinique.
3. Hypothèses alternatives concernant l’origine des cancers
La SMT, associée à l’hypothèse de l’initiation des cancers par des cellules initiatrices de tumeurs (Tumor Initiating Cells ou TICs) est devenue dominante en cancérologie. Toutefois, il est important de noter que Peter Nowell s’interrogeait sur la cause de la première modification néoplasique et ne favorisait pas l’idée qu’elle soit nécessairement mutationnelle. Dans l’article originel, il soulignait que
« Les conséquences biologiques de l’altération primaire peuvent être illustrées par des exemples… Les facteurs spécifiques qui produisent ces conséquences biologiques restent incertains. L’événement génétique spécifique qui les provoque est tout aussi obscur. L’absence de nouveaux produits géniques dans les cellules tumorales et la réversibilité de la transformation cellulaire dans certains systèmes de culture ont conduit certains chercheurs à suggérer que l’initiation tumorale implique généralement une altération de l’expression des gènes plutôt qu’une mutation structurelle. Il est clair que les altérations visibles de la structure des chromosomes ne sont pas essentielles au changement initial. La transformation peut se produire en culture tissulaire, et certaines tumeurs peuvent se développer in vivo sans anomalies cytogénétiques détectables. » [11]
La possibilité de réversion des tumeurs dans plusieurs circonstances est un argument fort suggérant que les mutations (dont la probabilité de réversion est extrêmement faible) ne seraient pas causales, du moins dans ces cas de réversion tumorale [18]. En outre, les études de séquençage à grande échelle ont révélé les limites du séquençage. Tout d’abord, les mutations observées ne suffisent pas à expliquer la transformation cancéreuse. Il est fréquent de retrouver ces mêmes mutations dans des tissus normaux. En effet, des cellules portant des mutations dites « driver », censées déclencher la tumorigenèse, sont aussi parfois présentes et abondantes dans des tissus sains [19]. De plus, le taux de mutation des cellules normales est comparable à celui mesuré dans plusieurs types de cancer [19]. Enfin, même dans le cas des oncogènes les plus fortement associés à l’émergence des cancers, comme l’oncogène Ras, la mutation oncogénique seule déclenche rarement des tumeurs ; ce sont plutôt des lésions tissulaires qui stimulent fortement la tumorigenèse en modifiant les états chromatiniens et la régulation de l’expression génique [17].
L’incapacité à expliquer l’émergence de toutes les tumeurs par des mutations d’oncogènes ou des gènes suppresseurs de tumeurs a stimulé l’élaboration d’hypothèses alternatives. Une hypothèse radicalement différente de la SMT est la « tissue organization field theory ou TOFT » [20, 21]. Selon cette théorie, les cancers n’ont pas nécessairement une origine clonale et mutationnelle. Ils seraient causés par des interactions anormales chroniques entre différentes composantes cellulaires d’un champ morphogénique donné à l’intérieur d’un tissu. Ces perturbations pourraient être provoquées par l’exposition à des carcinogènes ou à des perturbations physiologiques, induisant des changements stables d’expression génique, notamment par des altérations épigénétiques. Cette théorie fait écho à l’hypothèse selon laquelle des cellules peuvent, individuellement ou en groupe, changer de destinée et adopter des états fonctionnels alternatifs stables, sans mutations génétiques, similaires aux « vallées » du paysage épigénétique de Waddington (Figure 2 and Encart 2). mais cependant différentes des destinées stables des cellules normales, car résultant de perturbations stochastiques ou de perturbations externes comme une exposition à des agents carcinogènes [22, 23]. Une troisième hypothèse sur l’origine des cancers, appelée théorie de la « réversion évolutive », propose que les cellules cancéreuses atteindraient un état de prolifération incontrôlée en subissant une réversion vers des états similaires à ceux d’organismes unicellulaires, dont l’état par défaut est la prolifération. Les mécanismes d’inhibition proliférative, typiques des tissus soumis à un contrôle de leur taille, seraient ainsi perdus dans les cellules cancéreuses [24]. Ces états, représentant des étapes évolutives ancestrales, deviendraient stables et seraient donc à l’origine des cancers. .
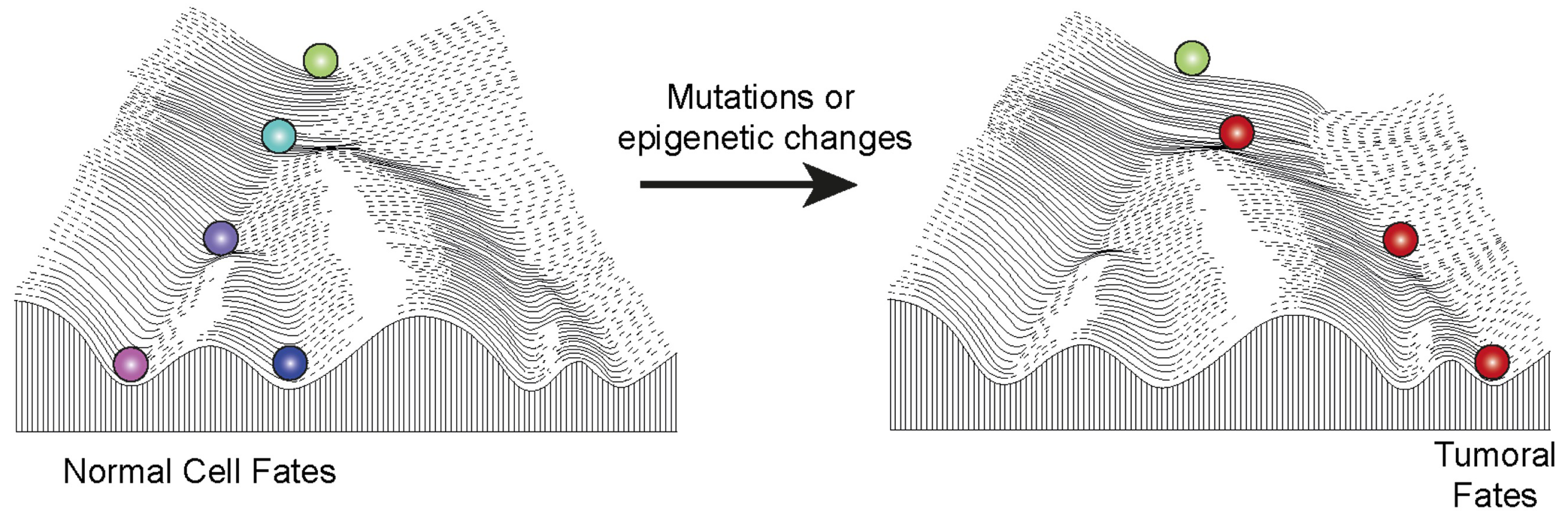
Le paysage épigénétique dans le développement normal ou dans le cancer. Le dessin illustre le célèbre paysage de Waddington, représentant une bille roulant sur une pente avec de multiples trajectoires possibles, déterminées par les collines et les vallées rencontrées sur son chemin. Ce paysage symbolise les multiples destins cellulaires pouvant naître d’une cellule pluripotente, telle que le zygote, et soutient l’hypothèse selon laquelle l’hérédité épigénétique contribue à la transmission stable des destins cellulaires. Les composants épigénétiques ou des expositions environnementales contribuent à façonner le paysage et conduisent à une variété de destins cellulaires. Dans le cadre d’un développement normal (à gauche), les cellules descendent la colline au cours de la différenciation afin d’acquérir des destins normaux. En cas de perturbation des composants épigénétiques, le paysage est altéré (à droite), forçant les cellules à emprunter un chemin aberrant pouvant mener au cancer. Masquer
Le paysage épigénétique dans le développement normal ou dans le cancer. Le dessin illustre le célèbre paysage de Waddington, représentant une bille roulant sur une pente avec de multiples trajectoires possibles, déterminées par les collines et les vallées rencontrées sur son ... Lire la suite
Enfin, le modèle des contraintes développementales, basé sur l’analyse extensive des données transcriptomiques à cellule unique de différents types de cancers comparés à des tissus sains, suggère que les tissus d’origine imposent aux cellules cancéreuses des contraintes de différentiation. Cela signifie que les cellules cancéreuses ne peuvent adopter qu’un nombre limité d’états cellulaires en fonction de leur tissu d’origine [25]. Ces observations présentent un intérêt considérable car elles permettent de mieux comprendre pourquoi chaque type de tissu ou cellule d’origine ne donnent lieu qu’à un nombre limité de types de cancers.
Encart 2 : Le paysage épigénétique de Waddington
Le célèbre paysage de Waddington (Figure 2) représente une bille dévalant une pente, pouvant suivre différentes trajectoires en fonction des vallées et des collines rencontrées sur son chemin. Cette illustration métaphorique illustre les divers destins cellulaires qu’une cellule, initialement représentée par le zygote, peut adopter au cours de son développement. Ce paysage est communément utilisé pour expliquer visuellement comment les mécanismes épigénétiques participent à la transmission stable des destins cellulaires, une fois qu’ils sont établis par des signaux intrinsèques et extrinsèques.
Les complexes Polycomb, grâce à leur capacité à réguler l’héritage épigénétique, peuvent jouer un rôle majeur dans le façonnement de ce paysage. Ils permettent aux cellules d’emprunter des trajectoires spécifiques, d’établir et de stabiliser différents états différenciés durant le développement normal. Cependant, des mutations ou des perturbations affectant le niveau d’activité des complexes Polycomb peuvent modifier ce paysage. Si ces perturbations sont suffisamment importantes, elles peuvent redessiner le paysage de manière à contraindre les cellules à suivre des trajectoires aberrantes mais intrinsèquement stables, favorisant ainsi la formation de cancers (Figure 2).
4. Facteurs épigénétiques et cancer
Bien que le rôle précis des mutations d’ADN et des changements épigénétiques, à différentes étapes, pour chaque typologie de cancer, soit encore à élucider, il est important de considérer que les facteurs impliqués dans l’héritage épigénétique contribuent significativement à la tumorigenèse [26, 27, 28]. Ces facteurs peuvent être regroupés en quatre grandes classes moléculaires.
Les ARN non codants appartiennent à plusieurs classes, chacune ayant ses propres mécanismes de production, de métabolisme et des fonctions biologiques spécifiques, souvent impliquées dans la tumorigenèse [29, 30]. Ce complexe moléculaire a la particularité de se fixer à la marque épigénétique 5-méthylcytosine, dans un contexte de séquence d’ADN riche en CG (mCpG), lorsque la cytosine est hémiméthylée. Ceci se produit notamment après la réplication de séquences complètement méthylées, car durant la réplication, chacun des brins d’ADN, qui porte des cytosines méthylées, est apparié à un nouveau brin portant des cytosines « naïves » et donc non-méthylées. La fixation du complexe DNMT1/UHRF1, suivie de la catalyse de la méthylation de la cytosine naïve, permet de rétablir l’état complètement méthylé, préservant ainsi la mémoire épigénétique de cette marque [31]. Cependant, la méthylation de l’ADN peut être modifiée, notamment par des processus d’oxydation impliquant des enzymes spécifiques appelées Ten-Eleven Translocation (TET), dont les altérations sont également associées au cancer [32].
Les ARN non codants appartiennent à plusieurs classes, chacune ayant ses propres mécanismes de production, de métabolisme et des fonctions biologiques spécifiques, souvent impliquées dans la tumorigenèse [33, 34, 35, 36]. Certains régulent des processus post-transcriptionnels, comme les microARN, tandis que d’autres influencent la régulation transcriptionnelle du génome. Ces ARN varient en taille : certains sont de petite taille (< 30 nucléotides), tandis que d’autres sont de grande taille (200 nucléotides ou plus, parfois plusieurs centaines de milliers de nucléotides). Selon le contexte moléculaire, ils peuvent jouer des rôles activateurs ou répresseurs. Ils peuvent affecter d’autres processus épigénétiques, tels que la méthylation de l’ADN ou les régulateurs de la composition ou de l’architecture de la chromatine [37, 38, 39].
L’hétérochromatine est une forme compacte et réprimée de la chromatine [40, 41, 42] qui contient de nombreuses protéines réprimant la transcription, ainsi qu’un grand nombre d’éléments d’ADN répétitifs. Elle peut former des domaines chromosomiques de plusieurs mégabases, recouverts d’une marque de triméthylation spécifique de l’histone H3K9 (H3K9m3), qui est déposée par les enzymes SUV39 et SETDB1. Cette marque est reconnue par des protéines hétérochromatiniennes, et leur fixation stimule leur activité catalytique. De plus, la marque H3K9m3 est associée à la protéine HP1 qui peut relier les nucléosomes adjacents. Ainsi, les composants de l’hétérochromatine peuvent à la fois déposer et se fixer à la marque H3K9me3, contribuant à la compaction de leur chromatine cible [43]. Cette présence dans le même complexe protéique de facteurs qui peuvent déposer une marque mais aussi la reconnaître peut stimuler d’une part la propagation de ces marques pour former de grands domaines chromatiniens et d’autre part peut contribuer à la stabilité de ces domaines et à leur transmission héréditaire [44]. En outre, les facteurs d’hétérochromatine collaborent avec d’autres facteurs chromatiniens pour transmettre l’hérédité épigénétique à travers les générations [43].
Enfin, les protéines Polycomb se regroupent principalement en deux classes de complexes : PRC2 et PRC1, responsables de la mise en place des marques H3K27me3 et H2AK119Ub, via EZH2 et RING1A/1B, respectivement [45]. Leur mode de recrutement dans des régions spécifiques du génome peut se faire par des protéines de liaison à l’ADN ou des ARN non codants. Comme dans le cas de l’hétérochromatine, les complexes Polycomb peuvent se fixer aux marques d’histone qu’ils déposent, facilitant la transmission de la mémoire épigénétique à travers les générations cellulaires et la meiose [46, 47, 48]. Leur action est également réversible, notamment grâce à protéines activatrices capables d’évincer les complexes Polycomb de la chromatine (telles que les protéines SWI/SNF) ou de remplacer les marques d’histone répressives par des marques activatrices (telles que les protéines des complexes COMPASS). Ces facteurs Polycomb, ainsi que SWI/SNF et COMPASS, sont fréquemment dérégulés ou mutés et contribuent à la tumorigenèse dans de nombreux types de cancers [49, 50].
Au cours des deux dernières décennies, de nombreuses études ont montré que ces différents facteurs épigénétiques – seuls et en combinaison avec d’autres composantes cellulaires et des facteurs environnementaux – jouent un rôle prépondérant dans les mécanismes de tumorigenèse [5, 51, 52]. Néanmoins, l’idée dominante reste que les mutations génétiques sont primordiales, tandis que l’épigénétique n’intervient qu’après les modifications de séquence pour accompagner ou aggraver le phénotype tumoral.
5. Initiation épigénétique de la tumorigenèse chez la drosophile
Les facteurs épigénétiques étant essentiels pour la régulation de l’expression de la majorité des gènes, il est pertinent de se demander si des changements épigénétiques peuvent initier un état tumoral (Figures 1 and 2). Toutefois, les changements épigénétiques étant souvent réversibles, la question est de savoir s’ils peuvent induire des états cellulaires aberrants suffisamment stables pour conduire à une pathologie comme le cancer. Les analyses génomiques et épigénomiques des cancers n’apportent pas de réponse concluante à cette question, car elles sont réalisées sur des tumeurs à des stades relativement avancés et révèlent généralement la présence de mutations ainsi que de changements épigénétiques, avec une grande hétérogénéité au sein des cellules tumorales. Par conséquent, il est très difficile d’identifier, parmi toutes ces variations, les changements spécifiques qui ont initié les premières cellules cancéreuses.
Pour distinguer les contributions épigénétiques des mutations génétiques, il est nécessaire d’induire une altération purement épigénétique, sans modification de la séquence d’ADN, et de tester si cette altération est suffisante pour initier un processus tumoral. Pour que cette étude soit concluante, elle doit exclure la possibilité que des changements génétiques survenus simultanément soient responsables du cancer. Les mutations survenant fréquemment, avec une ou plusieurs mutations apparaissant à chaque division cellulaire, il est nécessaire de séquencer et d’analyser les séquences des tumeurs ainsi générées. Idéalement, un tel système devrait permettre d’induire des cancers rapidement et de manière reproductible, afin de limiter les possibles contributions génétiques.
Un tel système a récemment été développé chez la Drosophile [53], un organisme modèle souvent utilisé pour l’étude des mécanismes fondamentaux de la transformation cellulaire. Ce modèle bénéficie d’une riche base de connaissances et d’outils génétiques, ainsi que d’une conservation évolutive des mécanismes d’initiation et de progression tumorale, incluant de nombreux oncogènes et gènes suppresseurs de tumeurs [54, 55, 56, 57].
Chez la Drosophile, les protéines Polycomb sont des suppresseurs de tumeurs qui exercent leur fonction en inhibant les voies de signalisation Notch, JNK et JAK-STAT [49, 58, 59, 60]. Des recherches ont montré que des tumeurs agressives apparaissent suite à une déplétion partielle de facteurs Polycomb durant le développement larvaire [61]. Ce système a été récemment exploité afin de tester l’effet d’une déplétion transitoire de ces protéines. Chez la Drosophile, il est possible de contrôler précisément dans quel tissu, à quel moment du développement, et pendant combien de temps une protéine de choix est supprimée, ceci grâce à un simple changement de température. A température dite permissive, la protéine est présente en quantité normale. A une température plus élevée (29 °C), un système d’interférence ARN est activé pour dépléter la protéine ciblée. Au moment souhaité, les Drosophiles sont à nouveau transférées à température permissive afin de restaurer l’expression normale du facteur qui avait été déplété.
De manière surprenante, des tumeurs agressives se développent après une déplétion de seulement 24 heures d’une protéine Polycomb [53]. Le séquençage de ces tumeurs a confirmé l’absence de mutations oncogéniques, prouvant que la tumorigenèse est bien initiée par la réduction transitoire d’un facteur Polycomb. Ceci signifie que des mouches dont le génome ne provoque pas de tumeurs à température permissive développent des tumeurs par simple changement transitoire de température. Ce changement transitoire n’induit pas de tumeur chez les Drosophiles contrôle qui ne contiennent pas le système de déplétion température-dépendant. Il est ainsi possible d’exclure que le changement de température soit la cause de la tumorigenèse. L’origine de ces tumeurs est donc purement épigénétique.
L’étude des mécanismes impliqués dans cette tumorigenèse épigénétique a montré que la déplétion de protéines Polycomb entraîne la dérégulation d’un grand nombre de gènes. Certains de ces gènes sont des cibles fixées directement par les protéines Polycomb dans leurs régions régulatrices. Cette fixation induit la condensation de la chromatine et réprime ainsi ces gènes. Lors de la perte de Polycomb, la transcription de ces gènes est alors activée. Une fois restaurée à la quantité normale, les protéines Polycomb se fixent généralement à nouveau sur leurs régions cibles. Cependant, la chromatine de certains gènes ne peut plus se condenser correctement, et un sous-ensemble de gènes continue à s’exprimer de manière anormale, conduisant à la tumorigenèse (Figure 1). La perte transitoire de facteurs Polycomb conduit donc à un état irréversible aberrant d’expression génique qui s’auto-maintient même après le retour des facteurs Polycomb, permettant aux cellules de devenir cancéreuses [53].
6. Initiation épigénétique de la tumorigenèse chez les mammifères
Si des facteurs épigénétiques peuvent être à l’origine de cancers chez la mouche, qu’en est-il chez les mammifères ? Des études de « preuve de concept » ont été réalisées chez la souris en utilisant des modèles de myélome multiple [62] et de lymphome des cellules [63]. Dans ces études, l’expression transitoire d’un oncogène (MafB pour le myélome multiple et Bcl6 pour le lymphome des cellules B) dans des cellules souches hématopoïétiques a conduit à l’apparition de tumeurs dans un délai de quelques semaines, avec des caractéristiques fortement similaires aux tumeurs humaines correspondantes. De manière intéressante, bien que l’expression de ces oncogènes disparaisse, ces cellules tumorales conservent un programme transcriptionnel tumoral stable. De plus, les cellules tumorales montrent des changements de méthylation de l’ADN. Dans le cas du lymphome des cellules B, les régions présentant des défauts de méthylation présentent un enrichissement de motifs reconnus par l’oncogène Bcl6, indiquant que les cellules tumorales ont développé un programme épigénétique stable suite à l’expression oncogénique transitoire.
L’ensemble de ces données suggère que des mécanismes épigénétiques peuvent initier la tumorigenèse, non seulement chez la drosophile, mais aussi chez la souris. Il est important de noter que ces études preuve de concept utilisent des mécanismes déclencheurs basés sur des actions transitoires (également définies comme « hit-and-run ») qui sont conçues au laboratoire. Cependant, des évènements de type « hit-and-run », fondés sur le principe de modifications transitoires de la fonction du génome, tels que des infections virales, peuvent également déclencher des tumeurs [64, 65], ce qui suggère que des événements naturels peuvent induire des processus de tumorigenèse.
7. Perspectives futures et biomédicales concernant le rôle de l’épigénétique dans le cancer
Même si les travaux expérimentaux sur les cancers d’origine épigénétique sont pertinents, plusieurs points restent à éclaircir. Tout d’abord, les mécanismes moléculaires précis par lesquels les cellules normales se transforment en cancéreuses restent à décrire. Ceci est vrai pour les études chez la souris tout comme celles chez la drosophile. Un suivi longitudinal détaillé pourrait fournir des informations majeures sur ces processus. Deuxièmement, il n’est pas encore clair si les cellules tumorales émergent de l’ensemble des cellules ayant subi une perturbation transitoire ou si des clones cellulaires spécifiques émergent suite à la perturbation. Des analyses approfondies à l’échelle de la cellule unique, ainsi qu’une analyse des séquences génomiques pour identifier d’éventuelles mutations dans des clones de cellules tumorales, pourraient aider à comprendre si des changements génétiques accompagnent, voire favorisent, l’action épigénétique dans le déclenchement de tumeurs, notamment chez la souris.
Il existe des tumeurs humaines pour lesquelles, malgré un séquençage approfondi, aucune mutation oncogénique n’a pu être identifiée. Parmi ces tumeurs, on trouve un sous-groupe des tumeurs stromales gastro-intestinales (GIST) [66, 67], ainsi que les hémangiomes infantiles [68]. Un autre exemple, qui a été étudié en détail, est l’épendymome de la fosse cérébrale postérieure (PFA) [69], un cancer pédiatrique du cerveau. Dans le PFA, des défauts de méthylation de l’ADN, une réduction de l’activité des protéines du complexe Polycomb PRC2 [70] et des altérations des repliements tridimensionnels du génome ont été observées dans les gènes associés [71, 72]. Les séquençages de ces tumeurs sont souvent limités à l’exome. Lorsque des séquences du génome entier sont obtenues, elles sont limitées à un petit nombre de patients et utilisent des techniques de séquences courtes qui ne permettent pas d’analyser les éléments répétés du génome. Dans le futur, des séquençages plus extensifs, notamment utilisant des techniques dites « long-read » qui permettent d’obtenir des lectures de plusieurs kilobases d’ADN, devraient permettre de mieux comprendre si des mutations d’ADN sont présentes dans ces tumeurs. Cependant, les données disponibles suggèrent que, pour ces types de cancer, des dérégulations de facteurs épigénétiques pourraient jouer un rôle déclencheur. De plus, même lorsque des mutations sont identifiées, elles pourraient être le résultat de la sélection de clones cellulaires successifs après la première transformation cellulaire. Les premières cellules tumorales pourraient donc être générées par des changements épigénétiques mais éliminées au cours du développement tumoral à un stade plus avancé. Il est donc important de poursuivre les études sur des modèles expérimentaux permettant d’approfondir notre compréhension des phénomènes moléculaires sous-jacents, ainsi que de se concentrer sur les cancers humains présentant peu ou pas de mutations oncogéniques, qui représentent des candidats de cancers épigénétiques.
Outre leur importance pour la compréhension fondamentale de la biologie du cancer, ces travaux pourraient avoir des conséquences sur les approches thérapeutiques de certains types de cancers. En particulier, des thérapies ciblant les facteurs épigénétiques commencent à être mises en œuvre. Ces traitements pourraient jouer un rôle grandissant à mesure que de nouvelles molécules seront développées pour cibler plus sélectivement des composantes épigénétiques spécifiques [73, 74, 75].
Une étude épidémiologique récente a clairement montré une forte augmentation de l’incidence des cancers diagnostiquées précocement (avant l’âge de 50 ans) [76]. Cette étude a identifié des facteurs de risque tels que l’alimentation, ainsi que la consommation d’alcool et de tabac, comme étant les facteurs de risque les plus corrélés à cette hausse [76]. Ces facteurs étant bien connus pour induire des modifications épigénétiques, il est envisageable que, plutôt qu’une augmentation massive des mutations, une altération des fonctions cellulaires et tissulaires, en partie due à des changements épigénétiques, soit à l’origine de cette augmentation de l’incidence des cancers précoces.
L’ensemble de ces données souligne la nécessité de décrypter non seulement les mécanismes de progression et de métastases du cancer, mais également ceux de son initiation. En effet, il est essentiel d’établir la chaîne complète des causes et conséquences qui, in fine, explique l’évolution de la maladie chez chaque patient. Cette compréhension nécessite une approche interdisciplinaire englobant des domaines aussi divers que les mathématiques et les sciences humaines et sociales [77]. Une telle approche pourrait permettre de non seulement améliorer le diagnostic et les approches thérapeutiques, mais aussi de développer des stratégies de prévention en se fondant sur les mécanismes moléculaires non mutagènes de la tumorigenèse [78, 79].
Glossaire
| DNMT1 | DNA methyltransferase 1 |
| UHRF1 | Ubiquitin-like, containing PHD and RING finger domains, 1 |
| mCpG | methyl-CpG |
| SUV39 | SUV39H1 histone lysine methyltransferase |
| SETDB1 | SET Domain Bifurcated Histone Lysine Methyltransferase 1 |
| HP1 | Heterochromatin protein 1 |
| PRC2 | Polycomb Repressive Complex 2 |
| PRC1 | Polycomb Repressive Complex 1 |
| EZH2 | Enhancer of Zeste Homolog 2 |
| RING1A/1B | Ring finger protein 1A/1B |
| H3K9me3 | Trimethylation of histone H3 on lysine 9 |
| H3K27me3 | Trimethylation of histone H3 on lysine 27 |
| H2AK119Ub | Monoubiquitinated lysine 119 of histone H2A |
| SWI/SNF | SWItch/Sucrose Non-Fermentable |
| COMPASS | COMplex of Proteins ASsociated with Set1 |
| JNK | Jun N-terminal Kinase |
| JAK-STAT | JAnus Kinase-Signal Transducer and Activator of Transcription |
| MafB | V-maf musculoaponeurotic fibrosarcoma oncogene homolog B |
| Bcl6 | B-cell lymphoma 6 |
| GIST | GastroIntestinal Stromal Tumors |
| PFA Ependymoma | Posterior Fossa type A Ependymoma |
Déclaration d’intérêts
Les auteurs ne travaillent pas, ne conseillent pas, ne possèdent pas de parts, ne reçoivent pas de fonds d’une organisation qui pourrait tirer profit de cet article, et n’ont déclaré aucune autre affiliation que leurs organismes de recherche.