

1 Introduction
The allylic substitution reaction (Fig. 1) is one of the most intensively studied catalytic reactions. Since the early reports by Tsuji 〚1–3〛 and Trost 〚4–6〛, it has become a powerful and versatile tool in synthetic organic chemistry. The reaction is compatible with a wide variety of allylic substrates as well as nucleophiles.
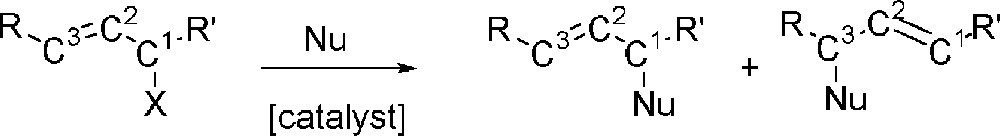
The allylic substitution reaction.
Recent research mainly focuses on asymmetric reaction variants 〚7〛 using often cyclic or linear 1,3-disubstituted acetates. The use of small substituents on the allyl moiety leads to less steric interaction between the ligand and the allyl group and consequently to a less efficient transfer of chiral information. Therefore, only ligands bearing the chiral centre in close proximity to the metal centre have been successful for such substrates 〚7, 8〛.
When mono-substituted allylic substrates such as cinnamyl (R=Ph, R’=H in Fig. 1) are used, regiocontrol is required prior to enantiocontrol. Palladium(bisphosphane) catalysts have been studied widely but show a preference for the formation of the linear product 〚9–11〛. Using mixed donor atom bidentate ligands, the regioselectivity can be steered 〚12–15〛. Recently it has been shown that catalysts based on metals such as tungsten 〚16〛, molybdenum 〚17〛, iridium 〚18–20〛 platinum 〚21, 22〛 and rhodium 〚23–25〛 show a preference for the chiral branched product, but the rate of the reaction is much lower than that observed for palladium.
Although the palladium-catalysed reaction is known to proceed via an η3-allyl complex, for the rhodium system an η1-allyl complex has been postulated as intermediate 〚23〛. The alkylation on the η1-allyl species will take place via an SN2’ attack of the nucleophile on the γ-carbon of the allyl moiety (Fig. 2). Alkylation of an allyl moiety with a substituent on the γ-carbon will yield the branched product.
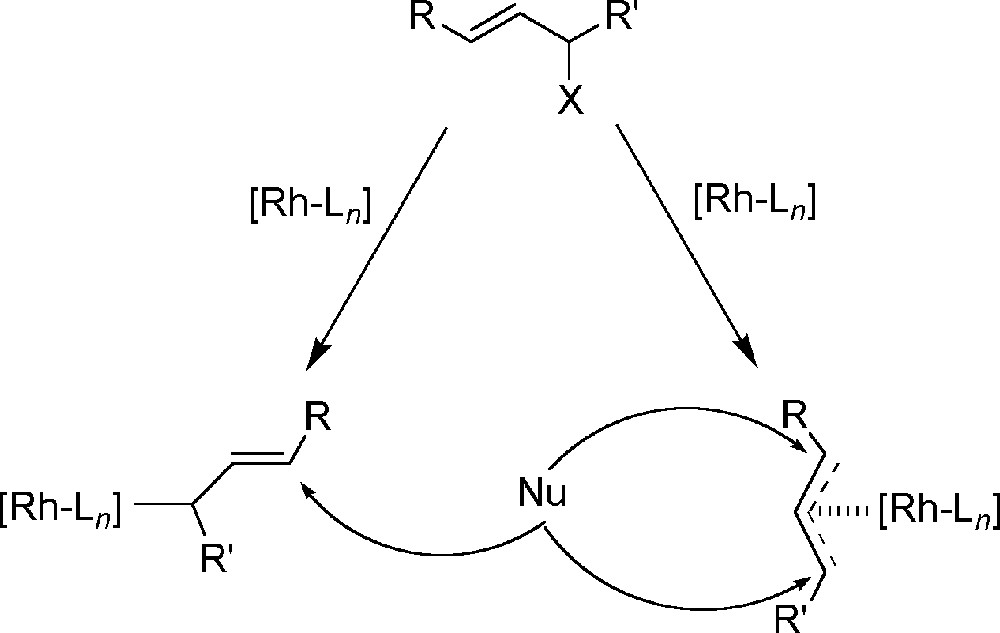
The postulated SN2’ mechanism for the rhodium-catalysed allylic alkylation via a Rh(η1-cinnamyl) species (left) and the alternative pathway via a Rh(η3-cinnamyl) species (right).
The rhodium catalysts in the reported alkylation studies were prepared in situ using an excess of monodentate phosphine ligands. For these systems, η1-allyl complexes have indeed been observed 〚26〛, but similar complexes bearing bidentate phosphine ligands have not been studied so far. In the late sixties, Vrieze 〚27〛 and the early eighties Fryzuk 〚28〛 have reported several studies concerning the synthesis and structure of allyl-rhodium complexes. Encouraged by their results we report here the synthesis and structure of novel (diphosphine)rhodium(allyl) complexes and evaluate their reactivity in the allylic alkylation reaction.
2 Results
2.1 Choice of ligands and allyl moieties
To study the effect of the bite angle of the ligand 〚29〛 on the geometry of (diphosphine)rhodium(allyl) complexes complex, four ligands having different preferred bite angles were used in this study (Fig. 3). Except for the ethanediyl bridged dppe (2), which enforces a relatively small bite angle and has a rather flexible backbone, the ligands are tri-aryl phosphines: o-dppb (1), which is a rigid ligand enforcing a small bite angle, DPEphos (3), which enforces an intermediate bite angle and has a flexible backbone and Xantphos (4), which enforces a large bite angle and has a more rigid backbone.

Ligands and complexes used in this study.
To study the effect of the substituent on the allyl moiety on the structure of the (diphosphine)rhodium(allyl) complexes, the small non-substituted C3H5 moiety (a) and the large phenyl substituted cinnamyl moiety (3-Ph–C3H4) (b) were used.
2.2 Synthesis
Starting from a RhI precursor, several routes were explored to synthesise (PP)RhIII(allyl) complexes. The most convenient and versatile route proceeds via oxidative addition of the appropriate allyl chloride to (PP)RhI(COD)Cl, resulting in the dichloro complexes (PP)RhIII(allyl)Cl2. The oxidative addition reaction proceeds smoothly for all complexes. Only for the DPEphos complex 3a, a small amount (< 5%) of an unidentified side product was formed that could not be removed.
2.3 Characterisation
An important issue in the characterisation of the newly synthesized Rh(allyl) complexes is the coordination mode (η1- or η3) of the allyl moiety. For palladium allyl complexes, the two coordination modes of the allyl moiety can be distinguished by several differences in the 1H-NMR spectra. For Pd(η3-allyl) complexes, the five hydrogens (Fig. 4) on the allyl moiety are non-equivalent in non-symmetrical complexes, whereas in symmetrical complexes {Hanti and Hanti’} and {Hsyn and Hsyn’} are equivalent.
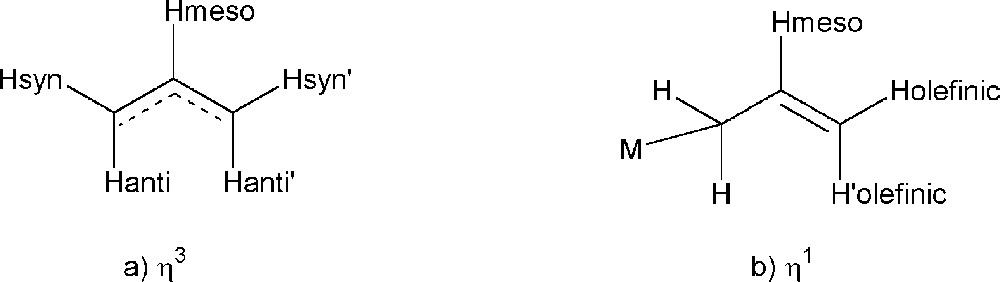
Allyl coordination modes, η3 (a) and η1 (b).
The anti hydrogens are closer to palladium and show a signal at lower ppm-value than the syn-hydrogens. In general, the signal for Hanti is typically found between 2 and 3.5 ppm, whereas the signal for Hsyn is mostly observed between 2.5 and 3.9 ppm. The signal of Hmeso almost always appears above 5 ppm, but below 6.5 ppm. In some cases, the η3-allyl moiety shows fluxional behaviour and Hanti and Hsyn become equivalent via a η3–η1–η3 rearrangement, which results in only one signal for the anti- and syn-hydrogens appearing at the averaged chemical shift.
The presence of a phenyl substituent at the allyl moiety (cinnamyl) disrupts the symmetry of the allyl group and all four protons show different signals. The phenyl can be oriented anti or syn with respect to Hmeso. The signals of Hanti and Hsyn, as compared to the C3H5 moiety, are in general shifted to a slightly higher ppm value. The chemical shift of the signal of the anti hydrogen next to the phenyl is found at lower field, because of the partial double bond character of the C2–C3 bond. The doublet may be observed even at higher chemical shift (between 5.9 and 6.8 ppm) than the signal of Hmeso, which is found between 5.8 and 6.6 ppm.
So far, only few (η1-C3H5)Pd complexes have been reported. The 1H-NMR spectrum resembles that of organic allyl compounds e.g. allyl chloride 〚30–32〛. The aliphatic CH2-unit appears as one signal; the olefinic signals are magnetically non-equivalent. The fine structure and the chemical shift of the signal of Hmeso resemble those found for Hmeso in η3-allyl complexes. The main difference, therefore, between an η3- and an η1-allyl is found in the fine structure on the signal of the olefinic CH2-unit. The signal consists of two double doublets around 5 ppm, whereas this is not observed for the η3-allyl complex.
The situation is slightly more complicated for cinnamyl complexes. In (triphos)Pd(η1-cinnamyl)Cl the cinnamyl is bonded to the metal via the CH2 unit, with a trans configuration of the allylic C=C bond. ((Triphos)Pd(η1-cinnamyl)Cl: 1H-NMR: δ = 2.3–2.6 (2 multiplets, 4H, backbone), 2.7 (m, 2H, Pd–CH2), 3.0–3.3 (m(ddd), 4H, backbone), 5.1 (dd, J1 = 15.3 Hz, J2 = 5.6. Hz, 1H, =CH–Ph), 5.8 (dt, J1(d) = 15.0 Hz, J2(t) = 6.3 Hz, 1H, Pd–CH2–C=CH–Ph), 7.0–7.8 (m, 30 H, aromatic H); 31P-NMR: δ = 47 (d, J = 48 Hz, 2P, –PPh2), 99 (J = 48 Hz, 1P, PPh2–(Ph)–PPh2); this palladium complex and other Pd(η1-cinnamyl) complexes are not reactive in the stoichiometric and the catalytic allylic alkylation; unpublished results). The 1H-NMR spectrum of these complexes shows one signal for the CH2 unit, a multiplet for Hmeso and a doublet for Holefinic (Holefinic is bonded to the terminal carbon atom). Thus, the spectra of Pd(η1-cinnamyl) compounds are expected to be quite similar to those of an η3-cinnamyl group, in which Hanti and Hsyn are equivalent or rapidly exchanging. Contrary to the η3-cinnamyl group, the η1-cinnamyl moiety does not show a phosphorus coupling on the olefinic hydrogen next to the phenyl group. Another tool to distinguish between the two coordination modes is the difference between the chemical shift of Hmeso and Holefinic. In the η3-allyl complex, the chemical shift of Holefinic is higher than that of Hmeso, whereas in the η1-allyl complex the chemical shift of the meso proton is higher than the olefinic proton.
We have used the similarities in the NMR spectra of the rhodium allyl complexes to the palladium complexes to assign the structure of the rhodium complexes (Tables 1 and 2). The interpretation of the 1H-NMR spectrum of the o-dppb cinnamyl complex 1b is hampered by the overlap of Holefinic and the aromatic hydrogens. Because of the similarities between the NMR spectra of 1b, 1a and 2b, the signals have been assigned to an η1-cinnamyl moiety. In addition, the Xantphos bearing complex, 4b, binds the cinnamyl in an η1-fashion. Both DPEphos modified complexes, 3a and 3b, have been identified as η3-allylic complexes.
Assignment of allyl coordination mode in (C3H5)Rh(PP)Cl2 complexes.
| Compound | PP ligand | Hapticity | Resonances in 31P-NMR | Remarks |
| 1a | o-dppb | η1 | 1 | |
| 2a | dppe | η1 | 1 | |
| 3a | DPEphos | η3 | 2 | all hydrogens non-equivalent |
| 4a | Xantphos | η3 | 1 | broad 31P-NMR signal |
Assignment of allyl coordination mode in (3-Ph-C3H4)Rh(PP)Cl2
| Compound | PP ligand | Hapticity | Resonances in 31P-NMR | Remarks |
| 1b | o-dppb | probably | 1 | Holefinic under aromatic signals |
| η1 | ||||
| 2b | dppe | η1 | 1 | |
| 3b | DPEphos | η3 | 2 | broad signals in 1H-NMR |
| 4b | Xantphos | η1 | 1 |
In most complexes, the two phosphorus atoms show only one signal in the 31P-NMR spectra, which is indicative of a symmetric complex geometry. Rhodium(III) can adopt several geometries: octahedral, square pyramidal, and trigonal bipyramidal. Although the coordination mode of the allyl is clearly identified by 1H-NMR, the geometry around the rhodium is not resolved. Since the chemical shift of 103Rh is known to be dependent on the geometry around the metal centre, attempts were made to measure the 103Rh-NMR signal. Unfortunately, the signals were too broad to be conclusive, even at low temperature.
After recrystallization of (Xantphos)Rh(Cl2)(η3-C3H5) 4a from CH2Cl2/hexane, crystals were obtained that were suitable for X-ray crystallography (Fig. 5). The structure of 4a is similar to the crystal structure of the analogous cationic (Xantphos)Pd(η3-C3H5)OTf 〚33〛. Geometrical data of both complexes are presented in Table 3.
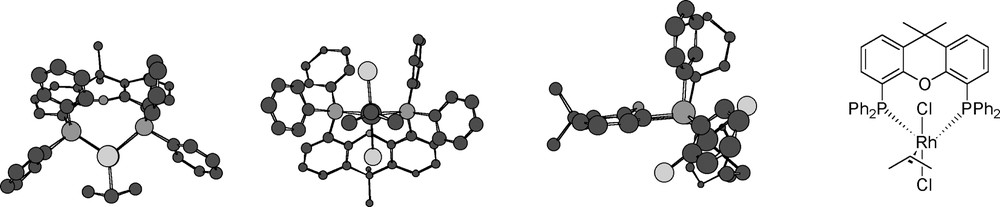
Crystal structure of complex 4a (three points of view and one schematic representation).
Selected geometrical data of the crystal structures of (Xantphos)Rh(Cl2)(η3-C3H5) (4a, Fig. 5) and (Xantphos)Pd(C3H5)+ OTf– 〚33〛. Distances in Å, angles in degrees (°). C1–C3 are the allylic carbon atoms, P1 is cis to C1, Cl1 is above in Fig. 4, Cl2 is below in Fig. 4.
| Rhodium | Palladium | |
| d(M–C1) | 2.232(11) | 2.17(1) |
| d(M–C2) | 2.186(14) | 2.16(1) |
| d(M–C3) | 2.241(11) | 2.17(1) |
| d(C1–C2) | 1.414(18) | 1.34(2) |
| d(C2–C3) | 1.442(18) | 1.34(2) |
| ∠(C1–C2–C3) | 124.9(14) | 120.4(15) |
| d(M–P1) | 2.394(2) | 2.372(2) |
| d(M–P2) | 2.437(2) | 2.372(2) |
| d(Rh–Cl1) | 2.351(3) | |
| d(Rh–Cl2) | 2.363(2) | |
| ∠(P–M–P) | 106.89(7) | 108.11(7) |
| ∠(Cl–Rh–Cl) | 177.44(10) | |
| angle between xanthene planes | 16.6571 | 27.352 |
| d(M–O) | 3.320(6) | 3.445(7) |
| ∠((P–M–P)–(C1–C2–C3)) | 58.9154 | 99.368 |
| d(C1–(P–M–P)) | –0.2723 (below) | +0.349 |
| d(C2–(P–M–P)) | +0.3136 | +1.008 |
| d(C3–(P–M–P)) | –0.2195 | +0.349 |
The rhodium complex has an octahedral structure, with the two chloride ligands in the axial positions, the two phosphine atoms cis to one another and the allyl in the equatorial plane bonded in an η3-fashion. In contrast to the palladium complex, the rhodium complex has no CS symmetry, probably because of the non-symmetric π–π interactions between the phenyl rings of the ligand. Compared to the palladium complex, the metal to allyl distance is larger and the C–C bonds in the allyl moiety are longer. Although the longer C–C bonds may suggest less olefinic character of the allyl moiety, the C–C–C angle of the allyl is larger. In addition, the angle between the P–M–P plane and the allyl C–C–C plane is much smaller. The coordination mode of the allyl group may thus indicate a decreased bonding and an increased backbonding interaction compared to the palladium complex. The P–Rh–P bite angle is similar to the calculated natural bite angle and to the bite angle found in other rhodium complexes of Xantphos derivatives 〚29〛.
The perpendicular stacking of the phenyl rings may explain the low chemical shift in the 1H-NMR spectrum (1.2 ppm) of one of the multiplet signals of the CH3 groups of the ligand. A different orientation of the phenyl rings causes a different folding of the Xanthene backbone and also a different ring current experienced by the methyl groups. Because of the large folding of the Xanthene backbone in the rhodium complex 4a, the difference between the endo and exo methyl group is smaller than in the analogous palladium complex.
2.4 Allylic alkylation
The rhodium allyl complexes 1b, 4a, 4b and the palladium analogue of 4b, (Xantphos)Pd(C3H5)+ have been used in the stoichiometric and catalytic alkylation using sodium diethyl 2-methylmalonate as the nucleophile. The results concerning the regioselectivity are shown in Tables 4 and 5. Since the rhodium-catalysed reactions proceed more slowly than the palladium-catalysed reactions, the former reactions are conducted using more concentrated reaction mixtures.
Stoichiometric alkylation of allyl complexes using sodium diethyl 2-methylmalonate as the nucleophile (100% yield, for reaction conditions: see experimental section).
| Complex | Branched (%)* | Linear (%)* | |
| 1b (o-dppb) (cinnamyl) | 88 | 12 | |
| 4b (Xantphos) (cinnamyl) | 23 | 77 | |
| 4a (Xantphos) (allyl) | 100 | ||
| Pd(Xantphos)(cinnamyl)OTf | 8 | 92 |
Catalytic allylic alkylation using sodium diethyl 2-methylmalonate as the nucleophile (for reaction conditions: see experimental section).
| Complex | TOFini* | % Conversion** | Branched (%)** | Linear (%)** |
| 4a (allyl) | 107 | 44 | 100 | |
| 4b (cinnamyl) | 15 | 13 | 21 | 79 |
The stoichiometric and the catalytic reactions show that probably both η3- and η1-allyl complexes are alkylated. In the stoichiometric alkylation, the regioselectivity for the formation of the branched product (nucleophilic attack at the substituted C3, see Fig. 1) is high for the o-dppb complex 1b (88%), whereas the Xantphos complex 4b shows a preference for the linear product and only 23% of the branched product is observed. Nevertheless, the selectivity to the branched product found using 4b is significantly higher than that found using its palladium analogue (Xantphos)(cinnamyl)OTf (8%). In the catalytic reaction of complex 4b, the same regioselectivity is found as in the stoichiometric reaction, suggesting that the reaction proceeds via the same pathway. Using the Xantphos rhodium complex, the reaction proceeds faster for allyl chloride than for cinnamyl chloride and at a rate similar to that of the palladium complex.
3 Discussion
3.1 Structures
For the rhodium allyl complexes with the smaller bite angle ligands (o-dppb and dppe), we obtained complexes in which the allyl moiety is coordinated in a η1-fashion. Although it can be expected that an octahedral structure is favoured for complexes having small bite angle ligands, such a coordination may put too much strain on the backbone of the ligand or may cause too much steric hindrance for the substituents on the phosphorus atom. Therefore, the geometry around the rhodium(o-dppb) and rhodium(dppe) are distorted. This distortion could explain the formation of η1-complexes for these ligands.
NMR spectroscopy shows that for DPEphos, both rhodium allyl complexes 3a and 3b are η3-allyl structures with most likely an octahedral geometry. The NMR studies show, that in contrast to the other ligands, the two phosphorus atoms of the C3H5 complex 3a are not equivalent, which indicates that the Rh-allyl bond is not symmetric. This has been observed before in crystal structures of cationic (DPEphos)Pd(allyl) complexes 〚33〛. It appeared that the backbone of the DPEphos ligand is folded, such that one of the aromatic rings of the backbone has a π–π interaction with one of the phenyl rings of the phosphorus atom that is bonded to the other backbone ring.
According to NMR spectroscopy, the Xantphos complexes 4a and 4b differ in structure and presumably also in the geometry around the rhodium. The crystal structure clearly shows an octahedral geometry for 4a, but for the η1-complex 4b calculations predict a square pyramidal complex 〚34〛. It is not clear whether the two phosphorus atoms are cis or trans to one another. A trans coordination of Xantphos, with a coordinated oxygen, has been observed in cationic palladium complexes 〚35〛 but seems less likely in a neutral rhodium complex. We therefore propose a cis coordination of the Xantphos ligand in complex 4b. The different coordination behaviour of C3H5 and cinnamyl to the rhodium is presumably caused by steric hindrance. In our previous work, it was described that a large cone angle of the ligand influences the coordination of the allyl group 〚33〛. A strong steric interaction between Xantphos and the cinnamyl moiety may therefore cause the different coordination behaviour observed in compounds 4a and 4b.
3.2 Allylic alkylation
It has been suggested that, in contrast to palladium, rhodium allyl complexes react via an SN2’ attack on the γ carbon of the η1-allyl moiety. We found that both η1- and η3-complexes react with sodium diethyl 2-methylmalonate to form the corresponding alkylated products indicating that both mechanisms are feasible for rhodium allyl complexes. Attack on the substituted γ-carbon of the η1-cinnamyl of o-dppb complex 1b results in a high regioselectivity for the branched product (88%). Remarkably, the alkylation of the analogous Xantphos η1-complex 4b results in the formation of only 23% of the branched product. This relatively low regioselectivity could be explained by a nucleophilic attack on an η3-allyl moiety (SN2) rather than substitution via an SN2’ mechanism. Possibly, complex 4b undergoes a rearrangement from η1 to η3 in the reaction mixture. The increased polarity of the solution and an interaction between the sodium of the nucleophile and a chloride of 4b may cause the formation of an intermediate rhodium complex with an enlarged Rh–Cl distance, which would enhance the possibility of η3-coordination of the cinnamyl group. The possibility of nucleophilic attack on a rhodium allyl complex is evidenced by the successful reaction between the η3-allyl complex 4a and malonate. If 4b reacts via the η3-structure, the higher selectivity for the branched product (23%) relative to the palladium analogue Pd(Xantphos)(cinnamyl)OTf (8%) can be explained by the large distortion of the Rh–cinnamyl bond (see above).
The catalytic alkylation of cinnamyl chloride using complex 4b shows the same regioselectivity as in the stoichiometric reaction, which indicates that the reactions proceed via the same intermediate. The alkylation of cinnamyl chloride using 4b proceeds relatively fast, even compared to palladium-catalysed allylic alkylation. The high reaction rate observed for complex could be explained by instability of an intermediate η3-cinnamyl complex and the large distortion of the Rh-cinnamyl bond. Furthermore, modelling studies 〚34〛 showed that for the η3-allyl complex, the backbonding interaction might be relatively small compared to the analogous palladium complex. We have shown that a smaller backbonding interaction enhances the reactivity towards nucleophilic attack 〚34, 36〛. The overall regioselectivity may therefore be a result of a competition between the SN2’ mechanism and the η3-mechanism. For η3-complexes, a larger bite angle of the ligand will result in an increase of the reactivity and the η3-mechanism may be favoured over the SN2’ mechanism.
4 Conclusion
We have synthesised and isolated a series of novel Cl2RhIII(diphosphine)allyl complexes. The coordination mode of the allyl moiety (η1 or η3) is highly dependent on the ligand and the substituents on the allyl moiety. The Rh(allyl) complexes react with malonate to form the alkylated product. The nature of the rhodium-allyl bond influences the mechanism of this reaction. In the allylic alkylation, an η1-complex reacts via the SN2’ mechanism whereas an η3-complex reacts via nucleophilic attack on the η3-allyl moiety. For palladium complexes, the η3-allyl square planar cationic geometry is favoured, whereas the crystal structure of 4a shows that neutral penta-coordinated complexes are formed for rhodium. The possibility for rhodium to adopt different geometries facilitates the formation of η1-allyl complexes. For palladium, η1-allyl complexes can only be formed when using tridentate ligands and coordinating counterions, and these η1-allyl species have shown to be non-reactive towards nucleophiles. In contrast, η1- and η3-allyl rhodium complexes described in this chapter react readily to form the alkylation product with a moderately high regioselectivity for the chiral, branched product.
5 Experimental section
5.1 General remarks
All reactions were performed in an atmosphere of argon unless stated otherwise. Dichloromethane was distilled under nitrogen atmosphere from P2O5; pentane, hexane, and toluene were distilled from sodium, THF, and diethyl ether from sodium/benzophenone. Bis-1,2-(diphenlylphoshino)-ethane and bis-1,2-(diphenylphoshino)-benzene were purchased from Aldrich and used as received. Xantphos and DPEphos were prepared according to a literature procedure 〚37〛. Allylchloride and cinnamylchloride were obtained from ACROS and used as received. 〚Rh(COD)Cl〛2 was synthesised according to a literature procedure 〚38〛.
The stoichiometric alkylation reactions were performed by adding an excess of sodium diethyl 2-methylmalonate (0.1 ml of a 0.5 M solution in THF) to a solution of 10 mg of the Rh-complex in 1 ml of THF. Reaction was instantaneous and after one minute, the mixture was worked up with water, filtered over silica, and analysed by GC.
The catalytic reactions were performed in THF (10 ml), using 0.5 mol% of catalyst (0.0050 mmol), 1.0 mmol of 3-Me-but-2-enyl acetate and 2.0 mmol of sodium diethyl 2-methylmalonate. The reaction was monitored by taking samples from the reaction mixture, which after quenching with wet ether, were analysed by GC using decane as the internal standard.
1H-, 31P{1H}- and 13C–NMR spectra were recorded at 300, 121 and 75 MHz respectively on a Varian FT NMR spectrometer. Variable temperature NMR experiments were performed on a Brucker DRX-300 FT NMR spectrometer equipped with a variable temperature unit. Chemical shifts are reported in δ units (ppm) and referenced to the residual deuterated solvent signal for 1H- and 13C–NMR spectroscopy, external H3PO4 (δ = 0 ppm) for 31P{1H}-NMR spectroscopy. The numbering scheme for the allyl moiety is shown in Fig. 4.
5.2 Rh(DPPE)(COD)Cl
16.1 mg of 〚Rh(COD)Cl〛2 (0.06 mmol) was dissolved in 5 ml of THF. To this solution, 26.5 mg of DPPE (0.06 mmol, 97%) was added while stirring. The solution was stirred for 30 min. The solvent was removed in vacuo and the solid was washed twice using 10 ml of pentane.
1H-NMR (CD3CN): δ = 8.0–7.0 (20H, m, aromatic protons DPPE), 4.1 (4H, bs, olefinic protons COD), 3.5 (2H, m, backbone protons DPPE), 2.3 (4H, bs, aliphatic protons COD), 2.1 (2H, m, backbone protons DPPE), 1.7 (4H, bs, aliphatic protons COD).
31P{1H}-NMR (CD3CN): δ = 63 (d, J(Rh,P) = 133 Hz).
5.3 Rh(Xanthphos)(COD)Cl
25 mg of 〚Rh(COD)Cl〛2 (0.05 mmol) was suspended in 5 ml of dry ether. To this suspension 58 mg of Xantphos was added as a finely ground solid. After addition, the solution was stirred for 30 min at room temperature, during which an orange solid precipitated. The orange solid was washed twice using 20 ml of ether. The product was dried under vacuum.
1H-NMR (toluene-D8): δ = 8.0–7.0 (26H, m, aromatic protons Xantphos), 4.5 (4H, bs, olefinic protons COD), 1.9 (4H, bs, aliphatic protons COD), 1.6 (6H, bs, methyl-protons Xantphos), 1.3 (4H, bs, aliphatic protons COD).
31P{1H}-NMR (toluene-D8): δ = 7.6 (d, 1J(Rh,P) = 91 Hz).
5.4 Rh(C3H5)(o-DPPB)Cl2 1a
50 mg of 〚Rh(COD)Cl〛2 (0.10 mmol) were suspended in 5 ml of toluene. To this suspension, 90.1 mg of o-di(diphenylphoshino)-benzene (0.20 mmol) in 15 ml of toluene was added dropwise.
After addition, the solution was stirred for 1 h, during which the product precipitated as an orange solid. 0.05 ml of allylchloride (0.65 mmol) was added. The mixture was stirred for another hour at room temperature, during which the colour of the suspension changed from orange to yellow. 15 ml of pentane were added to facilitate precipitation. The liquids were removed and the solid was washed twice using 20 ml of pentane. The solid was dried under vacuum.
1H-NMR (CDCl3): δ = 8.0–6.2 (24H, m, aromatic protons o-DPPB), 5.1 (1H, m, Hmeso), 5.0 (1H, dt, J(H,2H) = 4 Hz; J(H,H) = 14 Hz; H’olefinic), 4.9 (1H, dt, J(H,2H) = 3 Hz; J(H,H) = 6Hz, Holefinic), 2.6 (2H, d, J(H,H) = 8 Hz, H).
31P{1H}-NMR (CDCl3): δ = 62 (bd, J(Rh,P) = 124 Hz).
HR–MS (FAB): C33H29Cl2P2Rh+ requires m/z = 660.0176, found: 625.0491 (loss of one Cl).
5.5 Rh(C3H5)(DPPE)Cl2 2a
29 mg of Rh(DPPE)(COD)Cl (0.05 mmol) were dissolved in 5 ml of THF. To this suspension, 0.05 ml of allylchloride (0.65 mmol) was added dropwise. The mixture was stirred for 30 min at room temperature. The solvent was removed in vacuo and the product was washed twice using 10 ml of pentane. The yellow product was dried in vacuo.
1H-NMR (CDCl3): δ = 8.0–7.0 (20H, m, aromatic protons DPPE), 5.0 (1H, m, Hmeso), 4.8 (2H, m, Holefinic), 3.1 (2H, m, backbone protons DPPE), 2.9 (2H, bs, H), 2.5 ppm (2H, m, Backbone protons DPPE).
31P{1H}-NMR (CDCl3): δ = 66 (bd, J(Rh,P) = 105 Hz).
5.6 Rh(DPEphos)(C3H5)Cl2 3a
25 mg of 〚Rh(COD)Cl〛2 (0.05 mmol) were suspended in 5 ml of dry ether. 55 mg of DPEphos (0.10 mmol) was added as a finely ground solid to this suspension. After addition, the solution was stirred for 30 min at room temperature, during which a red solid precipitated. The solid was washed twice using 20 ml of ether. This solid was then suspended in 3 ml of THF. To this suspension, 0.05 ml of allylchloride (0.65 mmol, a large excess) was added dropwise. The mixture was stirred for 2 h, during which the colour of the solution changed from red to yellow and finally a yellow solid precipitated. The liquid was removed and the solid was washed twice using 20 ml of pentane. The solid was dried under vacuum.
1H-NMR (CDCl3): δ = 8.5–6.0 (aromatic protons DPEphos), 5.3 (1H, m, Hmeso), 4.5 (1H, dd, J(H,H) = 13 Hz, J(H,H) = 10 Hz, Hsyn), 4.2 (1H, dd, J(H,H) = 7 Hz, J(H,H) = 7 Hz, Hanti’), 3.1 (1H, d, J(H,H) = 7 Hz, Hsyn’), 3.0 (1H, d, J(H,H) = 12 Hz, Hanti).
31P{1H}-NMR (CDCl3): δ = 30 (1P, dd, J(Rh,P) = 115 Hz, J(P,P) = 9 Hz), 12 (1P, dd, J(Rh,P) = 144 Hz, J(P,P) = 9 Hz).
HR–MS (FAB): C39H33Cl2OP2Rh+ requires m/z = 752.0439, found: 717.0755 (loss of one Cl).
5.7 Rh(Xanthphos)(C3H5)Cl2 4a
42 mg of Rh(Xantphos)(COD)Cl (0.05 mmol) was suspended in 5 ml of toluene. 0.1 ml of allylchloride (1.3 mmol, a large excess) was added to this suspension. The reaction mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo, and the product was dried under high vacuum (5 × 10–5 mbar).
1H-NMR (CDCl3): δ = 8.0–7.0 (26H, m, aromatic H’s Xantphos), 5.5 (1H, p, J(H,4H) = 11 Hz, Hmeso), 3.6 (4H, bs, Hanti + Hsyn + Hsyn’ + Hanti’), 1.5 (6H, bs, CH3’s Xantphos).
31P{1H}-NMR (CDCl3) : δ = 5 (bs).
HR–MS (FAB): C42H37Cl2OP2Rh+ requires m/z = 792.0752, found: 757.1068 (loss of one Cl).
5.8 Rh(C3H4Ph)(o-DPPB)Cl2 1b
49.2 mg of 〚Rh(COD)Cl〛2 (0.10 mmol) was suspended in 5 ml of toluene. To this solution, 90.1 mg of o-di(diphenylphoshino)-benzene (0.20 mmol) in 15 ml of toluene was added dropwise. After addition, the solution was stirred for 1 h, during which the product precipitated as an orange solid. 0.10 ml of cinnamylchloride (1.3 mmol, a large excess) was added. The mixture was stirred for 2 h at room temperature. 15 ml of pentane were added to facilitate precipitation. The liquids were removed and the solid was washed twice using 20 ml of pentane. The solid was dried under vacuum.
1H-NMR (CDCl3): δ = 8.0–7.0 (30H, m, aromatic protons o-DPPB and phenyl cinnamyl-group), 5.6 (1H, dt, J(H,H) = 8 Hz, J(H,2H) = 11 Hz, Hmeso), 2.3 (2H, d, J(H,H) = 11 Hz, H).
31P{1H}-NMR (CDCl3): δ = 64 (d, J(Rh,P) = 164 Hz).
5.9 Rh(DPPE)(C3H4Ph)Cl2 2b
28 mg of Rh(DPPE)(CO)Cl (0.05 mmol) was dissolved in 5 ml of THF. To this solution, 0.05 ml of cinnamylchloride (0.65 mmol, a large excess) was added dropwise. The mixture was stirred for 30 min at room temperature. The solvent was removed in vacuo and the product was washed twice using 10 ml of pentane. The yellow product was dried in vacuo.
1H-NMR (CDCl3): δ = 8.0–7.0 (25H, m, aromatic protons DPPE and phenyl protons cinnamyl), 6.1 (1H, d, J(H,H = 25 Hz, H’olefinic), 5.7 (1H, dt, J(H,H) = 25 Hz, J(H,2H) = 8 Hz, Hmeso), 4.5 (2H, d, J(H,H) = 8 Hz, H), 3.3 (2H, m, backbone protons DPPE), 2.3 (2H, m, backbone protons DPPE).
31P{1H}-NMR (CDCl3): δ = 69 (d, J(Rh,P) = 145 Hz).
5.10 Rh(DPEphos)(C3H4Ph)Cl2 3b
25 mg of 〚Rh(COD)Cl〛2 (0.05 mmol) were suspended in 5 ml of dry ether. To this suspension, 55 mg of DPEphos (0.10 mmol) were added as a finely ground solid. After addition, the solution was stirred for 30 min at room temperature, during which a red solid precipitated. The solid was washed twice using 20 ml of ether. This solid was then suspended in 3 ml of toluene. To this suspension, 0.1 ml of cinnamylchloride (0.66 mmol, a large excess) was added. The solution was stirred at room temperature for 2 h. The product was precipitated using 20 ml of pentane. The liquids were removed, and the solid was washed twice, using 20 ml of pentane. The product was dried in vacuo.
1H-NMR (CDCl3): δ = 8.0–7.0 (33H, m, aromatic H’s Xantphos and phenyl cinnamyl-group), 6.1 (1H, bs, Hsyn), 5.8 (1H, m, Hmeso), 3.7 (1H, bs, Hsyn’), 3.4 (1H, bs, Hanti’).
31P{1H}-NMR (CDCl3): δ = 44 (1P, dd, J(Rh,P) = 180 Hz, J(P,P) = 15 Hz), 32 (1P, dd, J(Rh,P) = 134 Hz, J(P,P) = 15 Hz), 11 (0.1P, d, J(Rh,P) = 121 Hz, minor product).
5.11 Rh(Xantphos)(C3H4Ph)Cl2 4b
40 mg of Rh(Xantphos)(COD)Cl (0.05 mmol) were suspended in 5 ml of toluene. To this suspension, 0.1 ml of cinnamylchloride (0.66 mmol, a large excess) was added. The reaction mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo, and the yellow solid was washed twice using 10 ml of pentane. The product was dried under high vacuum (5 × 10–5 mbar).
1H-NMR (CDCl3): δ = 8.0–7.0 (31H, m, aromatic H’s Xantphos and phenyl cinnamyl-group), 6.7 (1H, d, J(H,H) = 16 Hz, Holefinic), 6.6 (1H, m, Hmeso), 4.4 (2H, bs, H), 1.7 (6H, s, methyl-protons Xantphos).
31P{1H}-NMR (CDCl3): δ = 16 (bd, J(Rh,P) = 117 Hz).
HR–MS (FAB): C48H4135Cl2OP2Rh+ requires m/z = 868.1065, found: 833.1384 (loss of one Cl).
5.12 Crystal structure determination of 4a
C42H37Cl2OP2Rh, Mw = 793.5, monoclinic, P21/c, a = 16.516(3) Å, b = 13.218(2) Å, c = 16.908(2) Å, β = 101.15(1)°, V = 3621.5(10) Å3, Z = 4, Dx = 1.46 g cm–3, λ(Cu Kα) = 1.5418Å, μ(Cu Kα) = 62.56 cm–1, F(000) = 1624, room temperature, final R = 0.074 for 5558 reflections.
A crystal with dimensions 0.15 × 0.20 × 0.45 mm approximately was used for data collection on an Enraf-Nonius CAD-4 diffractometer with graphite-monochromated Cu Kα radiation and ω–2θ scan. A total of 7453 unique reflections was measured within the range –20 ≤ h ≤ 20, 0 ≤ k ≤ 16, –21 ≤ l ≤ 0. Of these, 5558 were above the significance level of 4 σ(Fobs) and were treated as observed. The range of (sin θ)/λ was 0.031–0.627Å (2.7 ≤ θ ≤ 75.3°). Two reference reflections (〚2 1 〛, 〚 1 〛) were measured hourly and showed no decrease during the 132-h collecting duration. Unit-cell parameters were refined by a least squares fitting procedure using 23 reflections with 39.91 ≤ θ ≤ 41.00. Corrections for Lorentz and polarisation effects were applied. Absorption correction was performed with the program PLATON 〚39〛, following the method of North et al. 〚40〛, using Ψ-scans of five reflections, with coefficients in the range 0.356–0.954. The structure was solved by the PATTY option of the DIRDIF99 program system 〚41〛. The hydrogen atoms were calculated. Full-matrix least-squares refinement on F, anisotropic for the non-hydrogen atoms and isotropic for the hydrogen atoms keeping the latter fixed at their calculated positions with an atomic displacement parameter of U = 0.10 Å2, converged to R = 0.074, Rw = 0.081, (Δ/σ)max = 0.18, S = 1.07. A weighting scheme w = 〚15 + 0.01 × (σ(Fobs))2 + 0.01/(σ(Fobs))〛–1 was used. A final difference Fourier map revealed a residual electron density between –2.43 and 1.66 e Å–3 in the vicinity of the Rh. Scattering factors were taken from Cromer and Mann 〚42〛 and from the International Tables for X-ray Crystallography 〚43〛. The anomalous scattering of Rh, P and Cl was taken into account 〚44〛. All calculations were performed with XTAL3.7 〚45〛.