

1 Introduction
Transition metal cyanide chemistry is an area that experienced remarkable growth in recent years 〚1〛. Cyanide materials based on the well-known Prussian Blue compound have been revisited in light of their promising properties for an assortment of applications that include electronics 〚2〛, magnetism 〚3〛 and catalysis 〚4〛. In particular, the ability of the cyanide bridge to provide relatively strong and predictable magnetic coupling between transition metal centers has generated much attention in the field of molecular magnetism. This focus has led to tunable lightweight magnets that operate at high temperatures 〚5,6〛 and/or exhibit high coercivities 〚7–9〛.
A common trend in cyanide-based materials has been the emphasis directed towards the correlation between properties and structure. Many research groups have reduced the dimensionality of the cyanide phases to obtain crystalline compounds with a ‘divergent convergent’ methodology that involves reacting a hexacyanometalate anion with a coordinately unsaturated metal cationic complex 〚M(N–N)2(S)2〛n+ (N–N = bpy type ligand; S = facile leaving group such as solvent or halide). This strategy has yielded 2-D 〚10, 11〛 and 1-D 〚12–14〛 bimetallic networks whose magnetic properties reflect the reduced dimensionality of the materials. Most importantly, however, the dimensionality of some of these systems has reached the ‘zero-dimensional’ or molecular level. This has important consequences, because the potential for molecules to mimic solid-state properties of condensed phases are of great technological importance; e.g., certain high spin paramagnetic molecules 〚15, 16〛 behave as ‘single molecule magnets’ (SMM) 〚17–19〛.
The strategy of implementing convergent precursors has proven to be successful for the isolation of discrete molecules of various geometries, including squares 〚20〛 and cubes 〚21–23〛. However, this synthetic approach has limited control over chemical reactions, which are generally kinetically controlled processes. This often leads to the isolation of the most insoluble species from a reaction that involves equilibria between species. The insolubility of many of these species in aqueous media renders this chemistry even more difficult, since single crystals of sufficient quality for X-ray diffraction are not easily obtained.
In order to gain some insight into the different species formed in such reactions, we are using the diamagnetic and labile Zn(II) ion in mixed-metal cluster reactions. This approach can be advantageous, because it facilitates the isolation of more crystalline compounds, which may help with the future design of compounds with different metal ions. In this vein, Verdaguer and coworkers recently reported that various bimetallic networks can be obtained by reacting a cyanometallate precursor with a hydrated Zn(II) species 〚24, 25〛. In this paper, we provide details of the various molecules isolated from reactions that involve Zn(II) ions with capping ligands that help to keep the dimensionality of the products limited to zero-dimensional. This approach has led to the successful isolation of unprecedented molecular structures of various nuclearities and has provided important insight into the further development of high spin clusters.
2 Experimental Section
2.1 Materials
The starting materials ZnCl2·6 H2O, Zn(NO3)2·6 H2O, and K3Fe(CN)6 were purchased from Fisher Scientific and used as received. The reagents 2,2’-bipyridine (bpy) and 1,10-phenanthroline (phen) were purchased from Aldrich and used without further purification.
2.2 Syntheses
2.2.1 {〚Zn(bpy)2(OH2)〛2〚Fe(CN)6〛}Cl (1)·2 MeOH·12 H2O
A 10 mM methanol:water (1:1 v/v) solution of Zn(bpy)2Cl2 was prepared by adding 2 equiv of bpy to ZnC12·6 H2O. This solution was then combined with an equal volume of a 5 mM aqueous solution of K3Fe(CN)6 without stirring, to minimize the formation of a yellow precipitate. The solution was evaporated over a period of three days to yield X-ray quality yellow needles of 1.
2.2.2 {〚Zn(phen)2〛2〚Fe(CN)6〛2} (2)·〚Zn(bpy)3〛·32 H2O
A 5 mM aqueous solution of Zn(phen)2(NO3)2 was prepared by combining Zn(NO3)2·6 H2O with 2 equiv of 1,10-phenanthroline (phen). This solution was then mixed with a 10 mM aqueous solution of K3Fe(CN)6 without stirring, to minimize the initial formation of a yellow precipitate. The solution was evaporated slowly to yield light orange blocks of 2.
2.2.3 {〚Zn(bpy)2(H2O)〛〚Zn(bpy)2〛2〚Fe(CN)6〛2} (3)·23 H2O
A 10 mM solution of Zn(bpy)2C12 methanol:water (1:1 v/v) was prepared by adding 2 equiv of bpy to ZnC12·6 H2O. This solution was then carefully layered with a 5 mM aqueous solution of K3Fe(CN)6. X-ray-quality yellow needles of 2 formed over a period of one week.
2.2.4 {〚Zn(phen)2〛〚Fe(CN)6〛}2{〚Zn(phen)2〛〚Zn(phen)2(OH2)〛〚Fe(CN)6〛}2 (4)·4 MeOH·26 H2O
A 5 mM aqueous solution of Zn(phen)2(NO3)2 was prepared by combining Zn(NO3)2·6 H2O with 2 equiv of phen. This solution was then carefully layered over a 10 mM aqueous solution of K3Fe(CN)6 to yield an instantaneous yellow precipitate. After standing undisturbed for approximately four weeks, X-ray quality yellow needles of 4 had formed at the interface of the two solutions.
2.3 Single crystal X-ray diffraction studies
X-ray data were collected on a SMART 1K area detector diffractometer equipped with graphite monochromated Mo Kα radiation (λα = 0.710 73 Å). The frames were integrated in the Siemens SAINT software package 〚26〛, and the data were corrected for absorption using the SADABS program 〚27〛. The structures were solved using the direct-method program SIR97 〚28〛 and refined with SHELXL-97 〚29〛.
A yellow block of (1)·2 MeOH·12 H2O (0.40 × 0.05 × 0.03 mm3) was secured on the tip of a glass fiber with Dow Corning silicone grease and placed in a cold N2 (g) stream at 110(2) K. A total of 14 572 unique reflections were collected, although it should be pointed out that the dataset was very weak, due to the small size and poor quality of the crystal. The crystal system was determined to be triclinic, . All non-hydrogen atoms were located after successive Fourier difference maps, but due to the low data-to-parameter ratio, only the bpy atoms and metal centers were refined anisotropically. Hydrogen atoms were placed in calculated positions and their thermal parameters were fixed to be 20% larger than those of the atoms to which they are bound. The regions of the unit cell occupied by solvent are heavily disordered. The best fitting was obtained by assigning the electron density to water molecules with occupancy factors less than one (from 0.10 down to 1.0). The final refinement cycle was based on 14 572 reflections, with F0 > 2 σ(F0) and 457 parameters (R1 = 0.1141 and wR2 = 0.2727).
A light orange block of (2)·〚Zn(bpy)3〛·32 H2O (0.34 × 0.30 × 0.27 mm3) was secured on the tip of a glass fiber with Dow Corning silicone grease and placed in a cold N2 (g) stream at 110(2) K. A total of 22 360 unique reflections were collected. The crystal system was determined to be monoclinic, C2/c. All non-hydrogen atoms were located after successive Fourier difference maps; all non-solvent and non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and their thermal parameters were fixed to be 20% larger than those of the atoms to which they are bound. The regions of the unit cell occupied by solvent are heavily disordered. The best fitting was obtained by assigning electron density to water molecules with occupancy factors less than one (from 1.00 down to 0.30). The final refinement cycle was based on 11 899 reflections with F0 > 4 σ(F0) and 862 parameters (R1 = 0.0762 and wR2 = 0.1573).
A yellow needle of (3)·23 H2O (0.47 × 0.31 × 0.30 mm3) was secured on the tip of a glass fiber with Dow Corning silicone grease and placed in a cold N2 (g) stream at 110(2) K. A total of 37 484 unique reflections were collected. The crystal system was determined to be triclinic, . All non-hydrogen atoms were located after successive Fourier difference maps; all non-solvent and non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and their thermal parameters were fixed to be 20% larger than those of the atoms to which they are bound. The regions of the unit cell occupied by solvent are heavily disordered. The best fitting was obtained assigning the electron density to water molecules with occupancy factors less than one (from 1.00 down to 0.10). The final refinement cycle was based on 20 973 reflections with F0 > 4 σ(F0) and 1035 parameters (R1 = 0.0555 and wR2 = 0.1477).
A yellow needle of (4)·4 MeOH·26 H2O (0.12 × 0.04 × 0.04 mm3) was secured on the tip of a glass fiber with Dow Corning silicone grease and placed in a cold N2 (g) stream at 110(2) K. A total of 75 000 unique reflections were collected, although it should be pointed out that the dataset was very weak, due to the small size of the crystal. The crystal system was determined to be monoclinic, C2/c. Hydrogen atoms were placed in calculated positions and their thermal parameters were fixed to be 20% larger than those of the atoms to which they are bound. The final refinement cycle was based on 24 826 reflections with F0 > 4 σ(F0) and 1056 parameters (R1 = 0.0961 and wR2 = 0.1815).
3 Results and discussion
Crystal parameters are summarized in Table 1.
Crystal data and structural refinement parameters for compounds 1–4.
| Compound | (1)·Cl·2 MeOH·12 H2O | (2)·〚Zn(phen)3〛·32 H2O | (3)·23 H2O | (4)·4 MeOH·47 H2O |
| Emperical formula | C48H68ClFeN14O16Zn2 | C96H120Fe2N26O32Zn3 | C72H95Fe2N24O24Zn3 | C176H168N48O53Fe4Zn6 |
| Fw | 1319 | 2457 | 1973 | 4419 |
| space group | C2/c | C2c | ||
| a | 12.010(2) | 42.047(8) | 15.308(3) | 48.139(9) |
| b | 16.316(3) | 13.541(3) | 17.496(4) | 13.585(2) |
| c | 16.325(3) | 28.781(5) | 20.050(4) | 34.131(7) |
| α | 97.47(3) | 90 | 64.25(3) | 90 |
| β | 92.14(3) | 120.23(3) | 72.96(3) | 112.05(3) |
| γ | 92.43(3) | 90 | 88.50(3) | 90 |
| V (Å3) | 3166(1) | 14158(4) | 4592(I) | 20689(7) |
| Z | 2 | 4 | 5 | 4 |
| D (calcd) (g cm–3) | 1.279 | 1.407 | 1.392 | 1.395 |
| μ (Mo Kα) (Å) | 0.7169 | M169 | 0.7169 | 0.7169 |
| T (K) | 110(2) | 110(2) | 110(2) | 110(2) |
| R1, wR2 〚I > 3 σ (I)〛 | 0.1141, 0.2727 | 0.0762, 0.1573 | 0.0550, 0.1202 | 0.0961, 0.1815 |
3.1 Description of the structures
3.1.1 {〚Zn(bpy)2(OH2)〛2〚Fe(CN)6〛}Cl (1)·2 MeOH·12 H2O
The molecular structure of compound 1 reveals a cationic trinuclear entity of type {〚Zn(bpy)2(OH2)〛2〚Fe(CN)6〛}+ with Cl– as the counterion (Fig. 1). The compound consists of two trans-{Zn(bpy)2(OH2)}2+ units coordinated to a central 〚Fe(CN)6〛3+ unit (Zn–NC = 2.083 Å). Each trinuclear species contains a Δ and Λ ‘{Zn(bpy)2}2+’ unit with the vacant coordination site being occupied by a water molecule. These water molecules are stabilized by hydrogen bonding to the CN– ligand of the 〚Fe(CN)6〛3– unit, which leads to an O···N distance of 4.79 Å. The O atoms are also H-bonded to the Cl– ion (Cl···O = 2.81 Å). The water molecules that reside near and are H-bonded to the terminal cyanide ligands are disordered. The closest intermolecular contact in the structure is the π–π stacking interaction of the bpy ligands at a distance of 3.45 Å. The closest intermolecular M···M distance is 7.681 Å between the two Zn(II) centers.
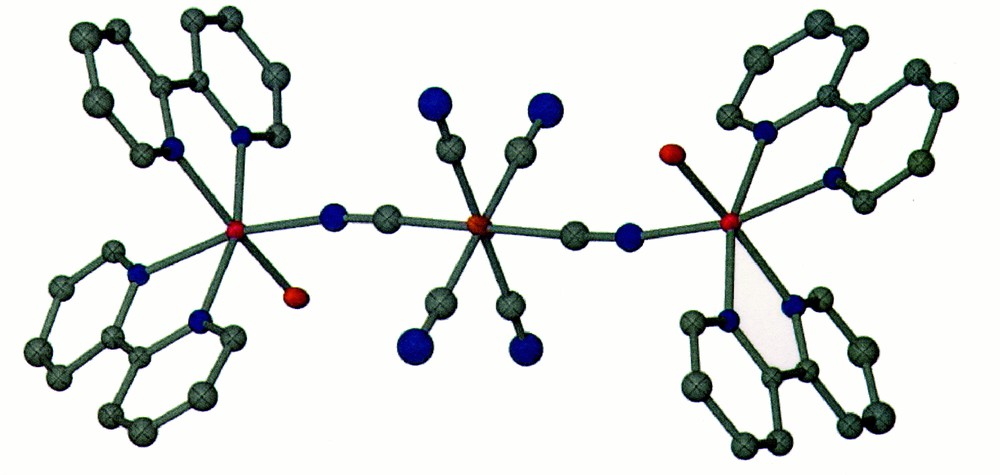
Color PovRay plot of the cluster {〚Zn(bpy)2(OH2)〛2〚Fe(CN)6〛}+ (1) (50% probability; Zn = pink, Fe = orange; N = blue; H atoms are omitted for clarity).
3.1.2 {〚Zn(phen)2〛2〚Fe(CN)6〛2}(2) 〚Zn(bpy)3〛·32 H2O
The crystal structure of compound 2 is that of a tetranuclear square dianion of the type {〚Zn(bpy)2〛2〚Fe(CN)6〛2}2– (Fig. 2) cocrystallized with a cationic 〚Zn(bpy)3〛2+ species. The 〚Fe(CN)6〛3– units reside at opposite vertices of the square motif with the other two vertices being occupied by a Δ and Λ {Zn(bpy)2}2+ unit. There are two Δ and Λ 〚Zn(bpy)3〛2+ isomers in each unit cell which leads to the centrosymmetric space group C2/c. The hexacyanometallate unit retains a more ideal geometry (Fe–C–N = 177.1(9)°, C–Fe–C = 88.7(4)°), but more distortion is evident in the coordination environment of the Zn(II) ion (Zn–N–C = 93.6(5)°, N–Zn–N = 93.6(4)°). This slight deviation from an ideal square geometry leaves an intramolecular Fe···Fe distance of 7.38 Å and Zn···Zn distance of 7.05 Å. The closest intermolecular contacts are between the phen ligands as a result of the π–π interactions (3.34 Å). The shortest intermolecular M···M contacts involve the 〚Zn(bpy)3〛2+ unit and the anionic square (Zn···Zn = 8.60 Å, Zn···Fe = 9.51 Å). There are disordered water molecules around the terminal cyanide ligands of the square that appear to be H-bonded.
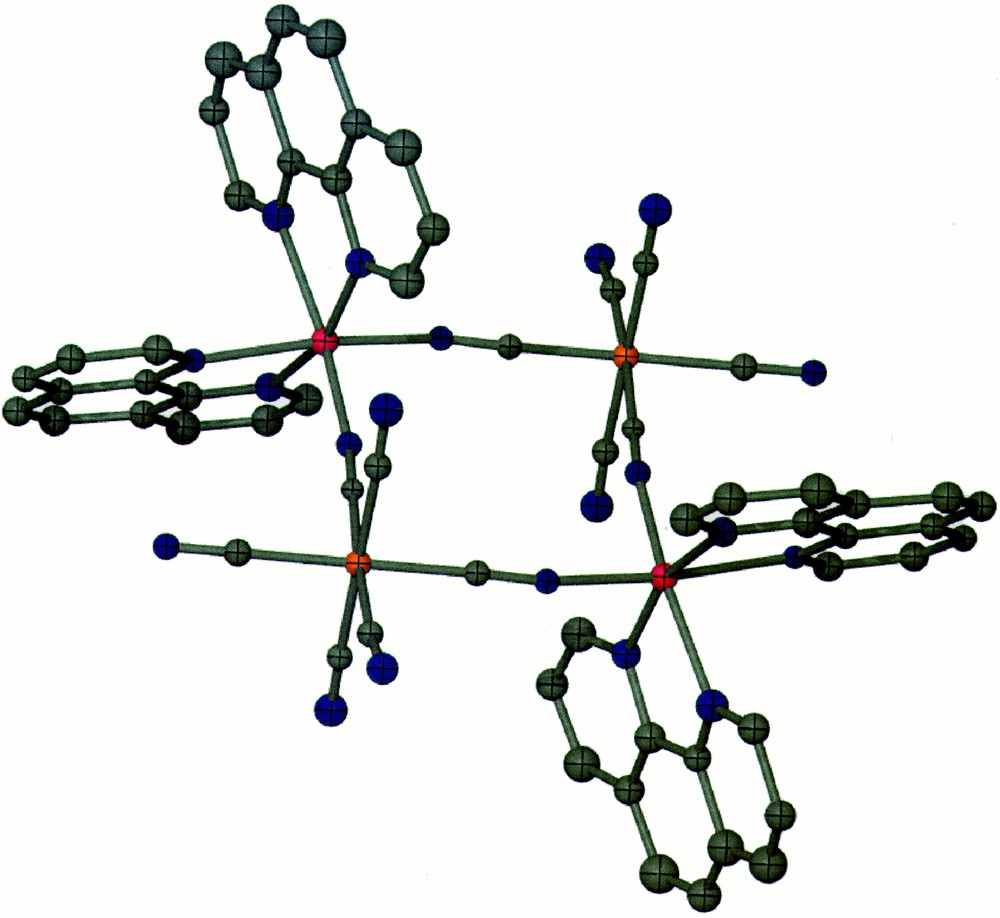
Color PovRay plot of the cluster {〚Zn(phen)2〛2〚Fe(CN)6〛2}2– (2) (50% probability; Zn = pink, Fe = orange; N = blue; H atoms are omitted for clarity).
3.1.3 {〚Zn(bpy)2(OH2)〛〚Zn(bpy)2〛2〚Fe(CN)6〛2} (3)·23 H2O
The molecular structure of compound 3 consists of neutral molecules of the type {〚Zn(bpy)2(OH2)〛〚Zn(bpy)2〛2〚Fe(CN)6〛2} (Fig. 3), whose basic geometry is that of an extended molecular square. The basic motif is {〚Zn(bpy)2〛2〚Fe(CN)6〛2}2–, with the four metal ions serving as corners and four bridging CN– ligands acting as edges. The two cis positions of the Zn complexes are coordinated to cis CN– ligands from independent hexacyanoferrate anions to yield a cyclic unit. Each square contains one Δ and one Λ {Zn(bpy)}2+ isomer. In addition, one of the 〚Fe(CN)6〛3– corners is bound to a third {Zn(bpy)2}2+ unit through one of the CN– units that lies in the plane of the square. The {Zn(bpy)2}2+ ‘appendage’ is coordinated to a water molecule that is perpendicular to the plane of the square unit (O···Zn distance = 2.15 Å). This mode of coordination leads to a bent angle for the Fe–C≡N–Zn bridge (C–N–Zn = 166.1(1)°), which is not unprecedented 〚30〛. In fact, angles as acute as C–N–Ni = 122° and C–N–Cu = 120° have been documented 〚31,32〛. As was observed for crystals of 1, disordered water molecules reside near the terminal cyanides, which implies the existence of some type of hydrogen bonding. The π–π interactions of 3.41 Å between bpy ligands of neighboring clusters are the shortest contacts, with the closest intercluster M···M distances being Zn···Zn’ = 7.77 Å and Fe···Zn = 8.44 Å.
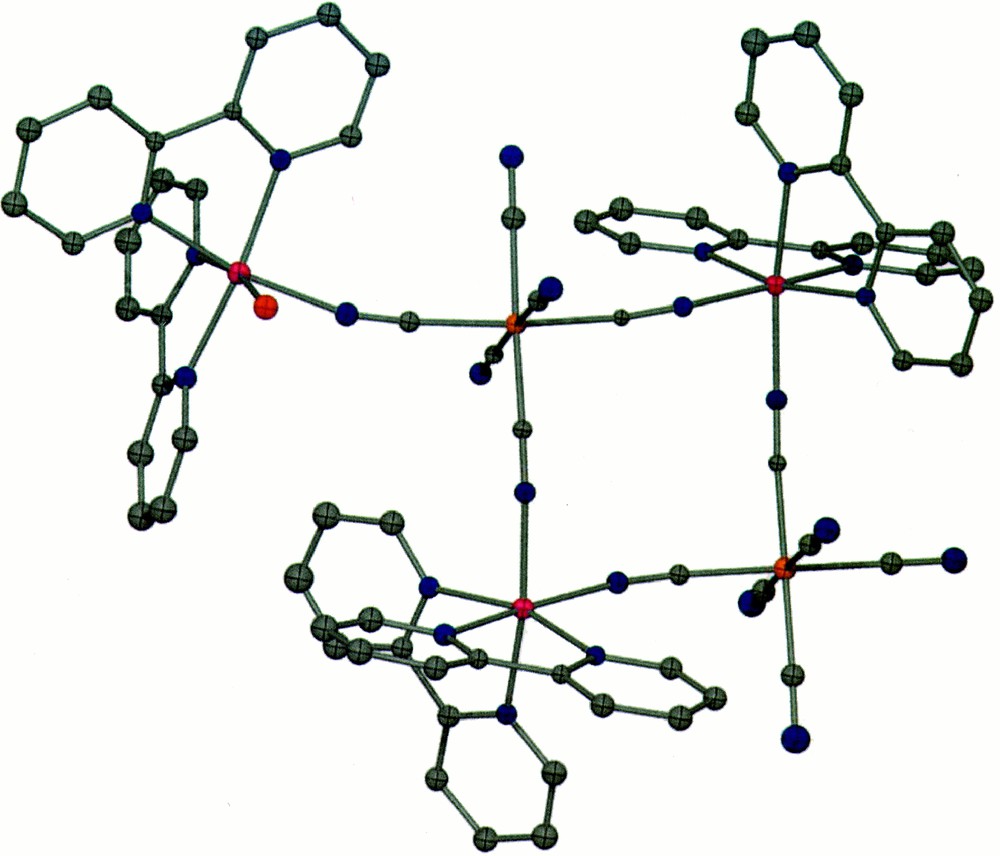
Color PovRay plot of the cluster 〚Zn(bpy)2(OH2)〛{〚Zn(bpy)2〛2〚Fe(CN)6〛2)} (3) (50% probability; Zn = pink, Fe = orange; N = blue; H atoms are omitted for clarity).
3.1.4 {〚Zn(phen)2〛〚Fe(CN)6〛}2{〚Zn(phen)2〛〚Zn(phen)2(OH2)〛〚Fe(CN)6〛}2 (4)·4 MeOH·26 H2O
The molecular structure of compound 4 consists of six {Zn(bpy)2}2+ units and four 〚Fe(CN)6〛3– units to form {〚Zn(phen)2〛〚Fe(CN)6〛}2{〚Zn(phen)2〛〚Zn(phen)2(OH2)〛〚Fe(CN)6〛}2 (Fig. 4). This highly unusual cluster can be reduced schematically to a dianionic central square unit (compound 2) with two monocationic trinuclear ‘appendages’ (compound 1) coordinated to the terminal cyanide ligands. This leads to a neutral decanuclear molecule of nearly planar geometry. The molecule contains three Δ and three Λ {Zn(bpy)2}2+ units and crystallizes in the centrosymmetric space group C2/c. While the square core is nearly ideal, the coordination of the trinuclear appendages to the CN– ligands gives rise to large deviations from linearity (Zn–N–Csquare = 163.1(3)°; Zn’–N–Cappendage = 146.8(5)°) The vacant coordination site of the terminal of the terminal {Zn(bpy)2}2+ unit is coordinated to a water molecule that is H-bonded to the cyanide ligand in the same place of the molecule. The π–π contact of 3.42 Å between bpy ligands or neighboring clusters are the closest contacts, whereas the closest intercluster M···M distances are Zn···Zn = 8.49 Å; Fe···Zn = 9.33 Å.
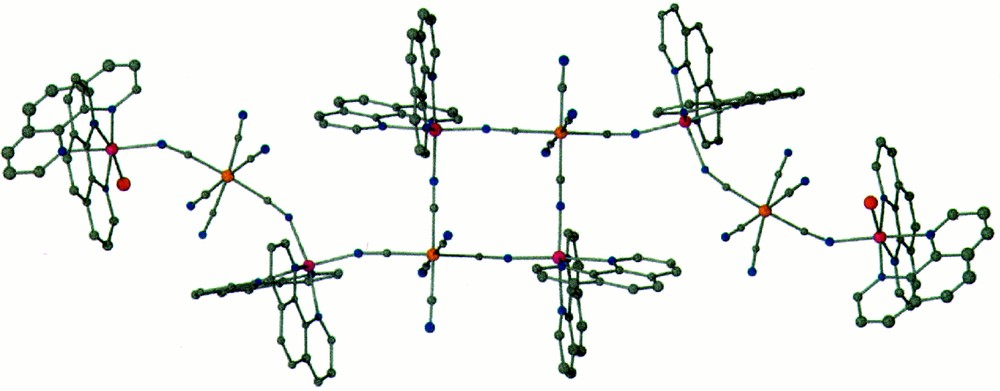
Color PovRay plot of the cluster {〚Zn(phen)2〛〚Fe(CN)6〛}2{〚Zn(phen)2〛〚Zn(phen)2(OH2)〛〚Fe(CN)6〛}2 (4) (50% probability; Zn = pink, Fe = orange; N = blue; H atoms are omitted for clarity).
3.2 Discussion
The results indicate that the solution chemistry of these compounds is dominated by equilibria between species of different nuclearities. The synthetic ability to displace these equilibria to attain the desired goal is key to obtain high yields. This study clearly indicates that the manipulation of experimental conditions leads to the isolation of a variety of products, as judged by single crystal X-ray diffraction. Most importantly, this investigation has revealed how subtle changes drastically affect the nature of the resulting products. Main factors to consider are the choice of ligand, solvent polarity, counterions and concentration. These factors contribute to the solubility of the molecular entities in solution and therefore affect the crystallization process. For example, neutral species are more likely to crystallize than ionic products due to lower solubility.
A perusal of the literature reveals that hexacyanometallate anions generally coordinate in a cis or fac orientation. As the current results indicate, however, both trans and cis coordination modes are possible. In contrast to compound 1, where two {Zn(bpy)2}2+ units coordinate in a trans fashion, the same units coordinate in a manner cis to the hexacyanometallate to form the molecular square in compound 2. In addition, both coordination modes are evident in compounds 3 and 4, which leads to the formation of unprecedented molecular geometries.
The general approach used in this study, namely the use of two cis capping ligands, has been shown to lead to the formation of molecular squares. The fact that isolation of the square is difficult in this case is attributed to the resulting anionic charge on the molecule, which renders it soluble in water. If one evaporates the reaction solution, however, it is possible to isolate the square along with a cocrystallized {Zn(bpy)3}2+ unit. If the square remains in solution for longer periods of time, it goes on to react with other cationic units, ultimately forming the insoluble compound 4. This demonstrates how the charge of these large molecules must be manipulated if they are to be isolated in high purity.
The synthetic strategies introduced herein may prove to be helpful in the isolation of high-spin cyanide clusters with an associated shape anisotropy based on a planar geometry. For example, it appears that hydrogen bonding of the coordinated water molecules with the in-plane terminal CN ligands plays a significant role in the geometry of the resulting product. Occupation of the vacant site by a water molecule in structures 3 and 4 prevents the formation of other molecular geometries (i.e. trigonal bipyramid geometry 〚33, 34〛) to afford planar structures of high nuclearity. Thus it seems reasonable to conclude that non-aqueous chemistry could lead to entirely different cluster geometries.
4 Conclusions
This study clearly demonstrates that subtle changes in experimental conditions can yield different products from the same cyanide-based precursors. An important lesson to be learned from these results is that elemental analysis is of little help due the polymorphism of these cyanide clusters, thus crystallographic characterization on single crystals is crucial. In light of the variety of clusters isolated in this study, it is naive to assume that the structure of single crystals that are obtained by slow diffusion reactions can be correlated to the magnetic behavior of a rapidly precipitated powder without further experimental evidence. Such assumptions will most likely result in the erroneous interpretation of magnetic data.
A more specific outcome of this study is that cyanide chemistry can yield planar, high-nuclearity clusters. This observation has positive ramifications for the field of SMM’s because molecular shape anisotropy, in addition to single ion anisotropy, is important for producing slow relaxation effects in magnetic molecules. It is also of interest to point out that the clusters in this study exhibit large intermolecular M···M distances (> 7 Å), with the closest intermolecular contacts being π–π stacking of the bpy or phen ligands, which is not an effective pathway for intercluster magnetic interactions. These crystallographic issues are encouraging, because if a ferromagnetic analogue of compound 4 with a negative ZFS parameter can be isolated with a large ground state spin value S, the molecule may exhibit SMM properties with relatively moderate blocking temperatures.
Finally, the present chemistry is being extrapolated; e.g., the 〚Fe(CN)6〛3– and {Zn(bpy)2}2+ building blocks are being replaced with 〚Mn(CN)6〛3– and {Co(bpy)2}2+, in an effort to yield clusters with large ground state values and a high degree of anisotropy, both of which are important ingredients for single molecule magnetic behavior.
Supplementary material
The crystallographic material for compounds 1–4 has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 IEZ, UK, as supplementary material CCDC 192744-192747, respectively, and can be obtained by contacting the CCDC (quoting the article details and the corresponding SUP number).
Acknowledgements
We thank the National Science Foundation for PI and NIRT grants (CHE-9906583 and DMR-0103455) and for the equipment grant for the CCD X-ray equipment (CHE-9807975). Support from the Welch Foundation and from a Telecommunications and Informatics Task Force (TITF) Grant from Texas A&M University is also gratefully acknowledged.