

1 Introduction
The pioneering work on the family of silica-based M41S mesoporous materials [1] with pore sizes from 20 to 100 Å opened a new era in shape-selective catalysis, guest-host chemistry, organic/inorganic composite materials and chromatographic separation [2]. Most of the research has been focused on hexagonal MCM-41 type material because of its easy availability and reproducibility. Less attention has been paid to the cubic MCM-48 system. In contrast to the one dimensional channel system of MCM-41, MCM-48 has a set of two, three dimensional interpenetrating pore systems and the channels of the mesopores can be used as the carrier for functional molecules or nanoparticles [3–5]. We reported recently the deposition of Cu-oxide, Zn-oxide and Cu/Zn-oxides within the MCM-48 pore system [6]. In addition, a number of reports have been published concerning nanosized metal oxide particles deposited in the mesopores of MCM-48 [7–9].
To find faster and economical synthesis routes for inorganic and organic materials has always been a challenging task, and the use of microwave heating for this purpose is now a subject of growing interest. Microwave synthesis might reduce the time of synthesis of standard preparations by almost two orders of magnitude. The microwave heating process works in a different way compared to conventional heating [10]. Dependent on the penetration of the synthesis medium, microwave energy can heat the entire object to crystallization temperature rapidly and uniformly resulting in homogeneous nucleation and shorter crystallization times compared to the hydrothermal method [11]. Moreover, this method is energy-efficient and economical. Microwave synthesis has been applied successfully for the synthesis of many zeolites and molecular sieves such as A, Y, ZSM-5, Beta, MCM-41, TS-1, aluminophosphates such as AlPO4-5, CoAPO-5, MgAPO-5, AlPO4-11 and large pore gallophosphate cloverite and GaPO4 [12–19]. Also mesoporous MCM-41 has been synthesized. So far no report on the microwave synthesis of MCM-48 has been published.
Conventionally, MCM-48 can be synthesized at 90 °C by heating the synthesis gel in an oven at autogenous pressure for 4 days. In this study we reported on the synthesis of MCM-48 using the microwave method and on its structural characterization with XRD, transmission electron microscopy (TEM) and N2 adsorption.
2 Experiments
2.1 Synthesis
MCM-48 was synthesized in polypropylene bottles under ambient pressure. 1 M NaOH solution (Baker) was taken in the bottle and cetyltrimethylammoniumbromide (CTABr, aldrich) was added along with water, under continuos stirring over a waterbath at 40–80 °C. Afterwards, tetraethoxysilane (TEOS, Merck) was added and the stirring was continued until the gel was homogeneous. The final molar gel composition of the synthesis mixture was TEOS: 0.15 CTABr: 0.5 NaOH: 80 H2O. The final gel was divided in two parts and poured in Teflon autoclaves for microwave heating. The microwave-oven used for this synthesis was a MDS-2000 system with a temperature controller as well as adjustable power output (maximum 650 W). The gel was allowed to heat at 100 °C for 1 and 2 h. The solid white products obtained were separated by filtration, washed with a mixture of H2O, Ethanol and HCl (molar ratio 18:1:2) and then dried at room temperature following a procedure similar as described in literature [20]. The surfactant inside the as-synthesized material was removed by calcination at 100 °C for 12 h then at 540 °C for 5 h.
2.2 Characterization
X-ray powder diffraction experiments were carried out using a Philips diffractometer with Cu–Kα radiation (λ = 1.5418 Å) in Bragg–Brentano geometry on flat plate sample holders. Single pulse 29Si MAS-NMR experiments (pulse width 5 μs) were performed with a Bruker ASX 400. The delay time between pulses was set to 300 and 60 s for as made and the calcined samples, respectively [6]. Spinning speed was set to about 3.5 kHz. Nitrogen adsorption isotherms were measured at the temperature of boiling nitrogen using an automatic Quantachrome NOVA 2000 Analyzer. Prior to the sorption experiment, the materials were dehydrated by evacuation under specific condition (300 °C, 2 h). The BET-surface area was calculated using the conventional BET equation [21]. The crystal size and morphology of the samples were monitored by SEM using a LEO-1530 Gemini microscope. TEM was performed using a Hitachi H-8100 instrument operating at 200 kV.
3 Results and discussion
The powder X-ray diffraction patterns for MCM-48 are shown in Fig. 1. The patterns of samples prepared by microwave hydrothermal method resembled to those prepared by the conventional, hydrothermal method. The domination 211 peak with a peak maximum at ~2.35° 2θ for Cu Kα radiation (d(211) ~ 37.6 Å) is the only resolved diffraction maximum. Indexing and analysis of peaks at higher angles was discarded because of to high experimental uncertainties. The half width of the 211 peak in the XRD pattern is wider in samples made using microwave heating, which might indicate less homogeneity of the mesoporous MCM-48 product. For mesoporous materials increasing peak half width in the diffraction pattern might be due to inhomogeneity or increased wall thickness. In addition, a mixture of different material types might cause partial signal overlap for the first peak, which will be discussed later. However, the XRD experiment can not reveal the reason for the ambiguity. In fact, because of the limited penetration depth of the microwave during the heating there is significant temperature difference between the liquid inside and the Teflon liner, which might lead to inhomogeneities in the process of formation. In order to differentiate MCM-48 from MCM-41 it is important to notice that for the first peak in the powder diffraction diagram at ~2.35° 2θ hexagonal indexing leads to the following 2θ positions for the 110 and 200 reflections of 4.1° and 4.7° 2θ, respectively. This is significantly different from the maxima in the range between 3.9° and 4.4° 2θ found in the sample synthesized using microwave heating which are typical for MCM-48 type materials. The calcination procedure was carried out in two steps. First, the sample was heated to 100 °C and annealed for 12 h then the temperature was increased up to 540 °C for 4 h. The XRD diagram of the calcined sample also shown in Fig. 1 reveals the characteristic shift of the first peak to lower d-values indicating the condensation process of the silicate species. The concomitant increase of peak intensity and half width makes the identification of the synthesis product difficult. The intensity increase of the main peak after calcination is a result of increased diffraction contrast between channel pore and wall (Fig. 1) [22]. This peak shift of ~0.7° 2θ is also typical for MCM-48 type material and was not observed for MCM-41 synthesized under similar conditions. TEM images (Fig. 2) reveal that parts of a mesoporous particles are well aligned with respect to the electron beam and show the periodicity of the MCM-48 silica framework, yet, some parts of the images show disorder and indicate wormhole like material which may be the reasons for the peak broadening, reflected in the X-ray diffraction diagram. For improving the structural periodicity, MCM-48 was kept at 50 °C for 4 days then calcined as described before. But no difference was noticed in the TEM images before and after annealing. There was not much difference in powder XRD patterns of the sample heated in the microwave for 1 and 2 h, which means 1 h heating was sufficient for successful synthesis. In 29Si MAS NMR (Fig. 3) the as made samples exhibited two broad resonances at –100 ppm for the Q3 environment and at –110 ppm for the Q4 environment, respectively. The hydrothermally prepared MCM-48 samples show the Q3/Q4 ratio significantly above 1, but interestingly this ratio is 1:1 for microwave prepared sample. The reason is not clear, but it may be one of the characteristic feature of the sample prepared using microwave heating. The ratio of Q3 to Q4 units decreased after calcination indicating the condensation of the silanol groups to form the more stable highly four-connected tetrahedral silicate network.
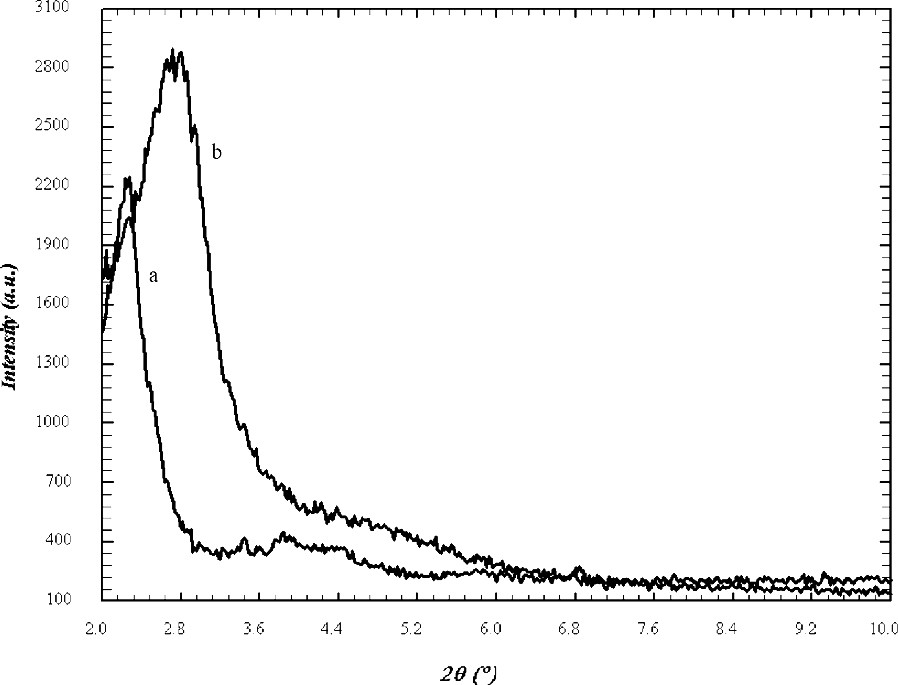
XRD patterns of (a) as made and (b) calcined MCM-48 samples prepared using microwave heating.

TEM image of a selected area of particles showing the co-existence of MCM-48 and wormhole silica framework along with the EDX analysis.
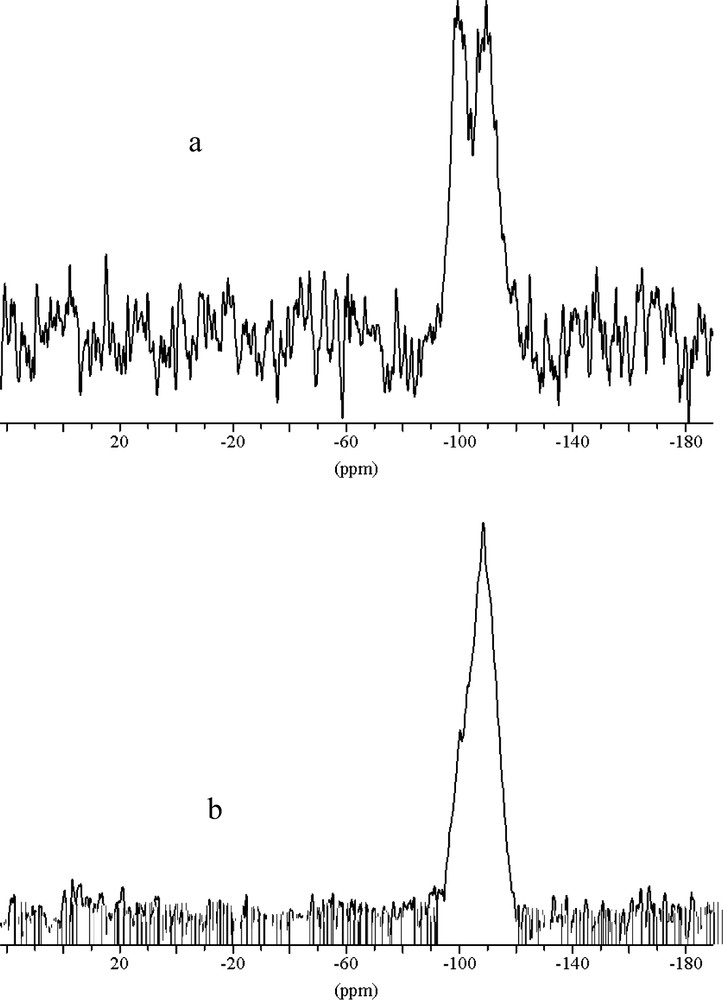
29Si NMR of (a) as made (b) calcined MCM-48 sample. The decrease of the high field Q3-signal after calcination indicates a higher degree of condensation as compared to MCM-48 made by the conventional hydrothermal method.
As shown in SEM (Fig. 4), the average size of the samples prepared by microwave method was about 1 μm, which was bigger than that prepared my normal hydrothermal method (0.4–0.5 μm). The morphology of the crystals is spherical and similar in both the methods.

SEM picture of a sample prepared in microwave oven showing sphere-like particles in the range between 1 and 2 μm.
The pore size distribution curve of the sample prepared using microwave heating is given in Fig. 5 and relevant data are summarized in Table 1. The sample shows similar behavior and properties as samples prepared with the conventional hydrothermal method. The surface area and pore diameter are in the range typical for MCM-48 type material [23], but slightly lower than those of samples prepared from similar composition using the conventional hydrothermal method. This can be explained considering the XRD patterns, 29Si MAS NMR spectroscopy and the TEM images. The peak in the powder XRD diagram of the sample made by microwave heating showed broad FWHM. Since the interference pattern in the PXRD experiment is the result of the periodic arrangement of slabs of high and low electron density in two dimensions and not of the periodic ordering of single atoms as scatterers, the wall thickness as contribution to the scattering from slabs of high electron density is reflected in the signal half width, i.e. the pore wall in the sample made using microwave heating might be thicker than those of samples synthesized by the conventional hydrothermal method. This is in agreement with the 29Si NMR spectrum with a higher content of Q4-linked silicate tetrahedra, which must be present in higher concentration in samples with thicker pore walls. The increased pore wall thickness leads to less surface area and pore volume in the material. As far as the wormhole structure is concerned, the pore diameter and the wall structure should be comparable with the major phase MCM-48 and should not take influence on the values derived from N2 adsorption measurements. However, the TEM results show that a fraction of the samples prepared via microwave route is not well ordered. Consequently, the experimental parameters used during the microwave treatment need to be optimized carefully in order to obtain perfectly ordered mesoporous materials. All attempts to synthesize pure phase MCM-48 using the microwave method failed so far.
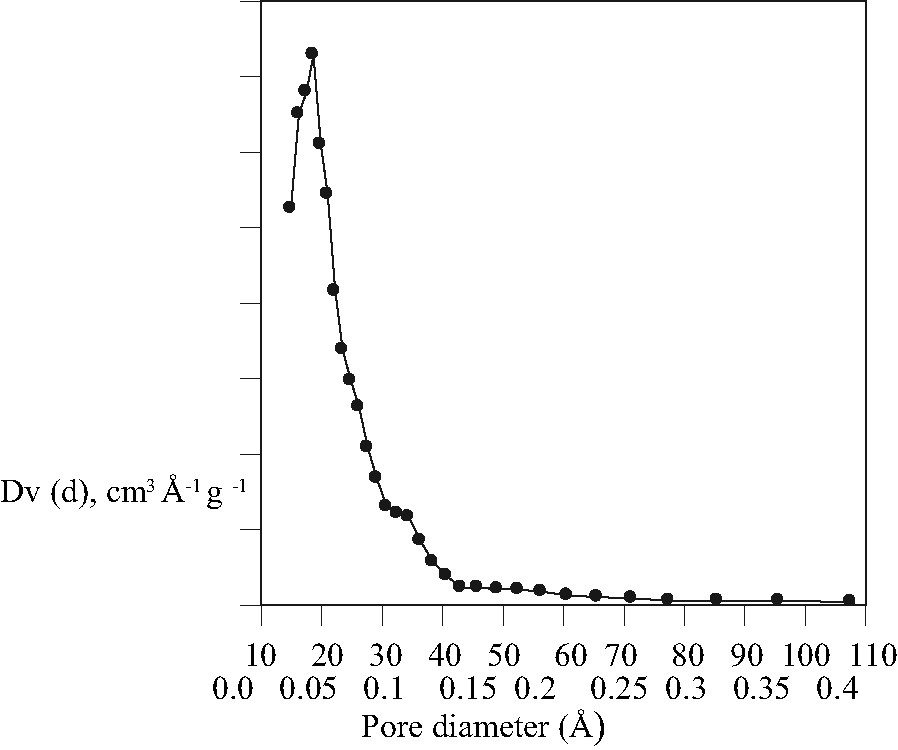
Pore size distribution curve of the MCM-48 sample synthesized using microwave heating.
Result of the N2 adsorption experiment
| Sample | BET-surface area (m2 g–1) | Total pore volume (BJH) (ccg–1) | Mean pore diameter (BJH) (nm) |
| MCM-48 (h) | 1257 | 0.93 | 2.5 |
| MCM-48 (m) | 835 | 0.70 | 2.0 |
4 Conclusion
The present study demonstrates the rapid synthesis of MCM-48 by the microwave method, at least as the major component in the reaction product. Much shorter time of crystallization has been achieved by this method compared to conventional synthesis. Although there is some structural disorder, mesoporous material with an intersecting channel system has been obtained. Some of the syntheses products yielded a better and more homogeneous products. Further improvement of the preparations is still needed to optimize the homogeneity of the synthesis product. However, the microwave heating method can be imagined as a time and energy-efficient route for obtaining MCM-48 type mesoporous materials.
Acknowledgments
Authors like to thank Technical Chemistry Department for measuring the N2 adsorption and Physical Chemistry 1 for the TEM measurement. This work has been supported by the Deutsche Forschungsgemeinschaft in the frame of the Sonderforschungsberich ‘Metal-support interactions in heterogenous catalysis’, SFB 558.