

1 Introduction
Block copolymers are used extensively to modify the properties of immiscible polymer blends. Usually, each block is chosen to be compatible with one of the homopolymer domains so that the that block copolymers localize at the interface between immiscible homopolymers with one block residing preferentially in each phase [1–6]. Localization of the block copolymer at the blend interface decreases the interfacial tension [7–10] and stabilizes the minor blend phase against coalescence [11], factors that decrease the phase size and subsequently improve mechanical properties such as toughness [12].
The entanglement of the two block copolymer sequences with the homopolymer phases across the interface can also lead to significant enhancement of the adhesion between homopolymers. The adhesion of polymer/polymer interfaces is often characterized by the fracture energy required to create a unit area of new surface at the interface. This energy is released from the elastic strain in the sample by propagating a crack at the interface and therefore is called the energy release rate or the fracture toughness, G. The effects of block copolymer addition on the fracture toughness of interfaces between two immiscible homopolymer glasses have been investigated extensively [13–15]. Fracture toughness generally increases upon addition of block copolymers, and in favorable cases reaches values similar to the cohesive strength of one of the homopolymers. For relatively low copolymer molecular weights the interface was found to fail by chain pull-out without crazing. In this process, one of the blocks is progressively pulled out from the respective homopolymer domain upon fracture. The fracture toughness in the chain pull-out regime was found to increase linearly with the areal chain density (Σ) of block copolymers, and scaled roughly with N2, where N is degree of polymerization of the pulled-out block. The dependence on Σ and N agree with the model of chain pull-out proposed by Xu et al. [16].
The fracture toughness of interphases between rubbery and glassy polymers was initially studied by Brown and Creton [17,18] using the so-called JKR technique [19]. They examined the adhesion between polyisoprene (PI) elastomer lenses and a glassy polystyrene (PS) substrate, and how the adhesion changed when thin layers of P(S-b-I) diblock copolymer were deposited at the interface. The threshold toughness increased linearly with the areal density of the copolymer chains, in agreement with the prediction of the weak junction models of chain pullout [20–22]. However, the increase of G0 with molecular weight did not show the linear dependence on N expected from the models.
Leger and coworkers [23–25] studied the adhesion of silicone elastomers to silicon substrates modified with poly(dimethyl siloxane) brushes. Adhesion tests exhibited a maximum in the adhesion energy with change in the areal density of the PDMS brushes. This was explained by the increasing difficulty in penetration of the brush molecules into the elastomer gel with increase in brush areal density. The process of brush interpenetration into the gel was found to be an exceeding slow process, taking several days to weeks to come to equilibrium.
The adhesion enhancement offered by block copolymer addition is also affected by chemical grafting reactions between the copolymer and the polymeric substrate. Reichert and Brown [26] demonstrated that, for the P(S-b-I) system, greater adhesion values could be obtained by crosslinking the polyisoprene block copolymer sequence into the PI elastomer. A copolymer that is covalently linked to the elastomer phase must fail by chain scission and therefore should serve as a good model system for examining the Lake–Thomas [27] theory of failure in crosslinked networks.
Interfacial reactions/interactions are often sited as an origin of the adhesion hysteresis phenomenon observed in many studies of PDMS gel adhesion. Silberzan et al. [28] attributed hysteresis, upon loading of two PDMS hemispheres, to hydrogen bonding between SiOH groups on the two surfaces. Perutz et al. [29] observed adhesion hysteresis between PDMS networks, that had been deliberately hydrolyzed by exposure to 0.1 M HCl aqueous solutions. Hysteresis was found to increase with exposure time. When the silanol end groups were replaced by reaction with hexamethyldisilazane (which replaces the end-group with a trimethylsilyl group), the hysteresis was reduced by up to 80%. In contrast, Emerson et al. [30] demonstrated that the presence of SiH groups was not required for hysteresis to occur between toluene-extracted hemispheres. Unreacted SiH groups were removed by reaction with ethylene gas, but hysteresis was still observed. In this case it was argued that hysteresis resulted from the entanglement of chains across the interface.
Adhesion hysteresis has also been observed for PDMS in contact with unlike substrates. Kim et al. [31] observed adhesion hysteresis when PDMS hemispheres were contacted with a variety of self-assembled monolayers on silica surfaces. Hysteresis was observed for PDMS hemispheres in contact with bare silicon wafers. Less hysteresis was observed for a surface consisting of carboxylic acid groups, an even smaller amount of hysteresis was observed for a surface functionalized with biphenyl groups, and virtually no hysteresis was observed for a surface modified with a perfluorocarbon. These differences were explained in terms of specific interactions at the surface with the high degree of hysteresis observed against bare silica substrates resulting from hydrogen bonding. Choi et al. [32] examined the effect of increased acidity on the adhesion between PDMS hemispheres and various OH functionalized self-assembled monolayers on gold substrates. Adhesion increased with increasing acidity and density of OH groups, though the relationship was highly non-linear.
The present paper considers the use of a reactive copolymer, with one glassy block and a second elastomeric block terminated with a reactive functional group, to reinforce the interface between a glassy homopolymer and an elastomer. The block copolymer can mechanically bridge the polymer blend interface by reaction of the functional group on the end of the elastomeric copolymer block with residual functionality in the elastomer gel and entanglement of the glassy polymer block with the glassy homopolymer. The system selected for study is a silane-terminated poly (styrene-b-dimethyl siloxane) diblock copolymer deposited at the interface between a poly (dimethyl siloxane) (PDMS) elastomeric gel and a glassy PS substrate. We previously reported that these surface-active ω-functional diblock copolymers can be used to create smart surfaces with selective adhesion properties [33]. Polystyrene surfaces modified with the silane terminated block copolymers selectively promoted adhesion to PDMS substrates while surfaces modified with carboxylic acid terminated block copolymers promoted adhesion selectively toward poly (methyl methacrylate) substrates. We have also shown that the occurrence of an interfacial reaction in this system is responsible for the adhesion hysteresis observed in finite rate adhesion tests [34] performed by the JKR technique. The purpose of this paper is to investigate the mechanism of adhesion promotion by these reactive, end-functional block copolymer brushes when applied to the interface between a PDMS gel and a PS substrate.
2 Experimental
2.1 Materials
End-functional poly(styrene-b-dimethyl siloxane) diblock copolymers in which the siloxane block was terminated with a reactive silane group were synthesized by standard anionic methods using sec-butyl lithium as the initiator and dimethyl chlorosilane as the terminating agent. This procedure provides reactive silane functionality at the end of the PDMS block. Characteristics of the polymers employed in this study are listed in Table 1. In view of the difficulties in determining absolute molecular weights and polydispersities of block copolymers, characterizations were performed by multiple groups using size exclusion chromatography (SEC) calibrated with polystyrene standards and by simultaneous size exclusion chromatography laser light scattering (SEC/LS). Since absolute molecular weight data was not available for all of the samples we have not attempted any type of universal calibration of the molecular weight. Instead, we employ the GPC molecular weight when examining results, recognizing that these values are about 20% below the absolute values.
Characteristics of Block Copolymers (Molecular weights are in units of kDalton)
|
| UV |
|
|
| dn/dc | ||||
| Mtot | MPDMS | Mw/Mn | ωPDMS | Mtot | MPDMS | MPDMS | Mtot | MPDMS | ϕPDMS | |
| P(S-b-DMS)47 K | 109 | 54 | 1.10 | 0.495 | 68 | 40 | 47 | 102 | 57 | 0.44 |
| P(S-b-DMS)54 K | 91 | 55 | 1.08 | 0.604 | 80 | 52 | 54 | 108 | 68 | 0.37 |
| P(S-b-DMS)60 K | 101 | 58 | 1.08 | 0.574 | 103 | 63 | 60 | 139 | 81 | 0.42 |
| P(S-b-DMS)65 K | 110 | 62 | 1.08 | 0.563 | 108 | 68 | 65 | n.a. | n.a. | n.a. |
The sample nomenclature adopted for the block copolymers is P(S-b-DMS)XK, where X is the SEC average molecular weight of the PDMS block. Thus the block copolymer with SEC average molecular weight of 65 000 is denoted as P(S-b-DMS)65 K. The PS composition of each diblock copolymer was obtained by Ultraviolet–Visible spectroscopy (UV) and through analysis of the refractive index increment. The end-functional PS homopolymer, PS–Si–(O–CH2–CH3)3 with Mn = 160 000 g/mol and Mw/Mn = 1.05, was synthesized anionically by terminating a living polystyryl-lithium anion with trimethoxychlorosilane. α,ω-Divinyl-terminated PDMS with a viscosity of 100 cs (MW = 6000 g/mol), purchased from Petrarch Systems, was used as the starting materials for the lenses. The crosslinking agent (182 cure agent, MW≈2000 g/mol) for the lenses, provided by Dow Chemicals Co., was poly(methylhydrosiloxane-co-dimethylsiloxane) copolymer containing 30–35% methylhydrosiloxane. The catalyst for the crosslinking reaction was PC075 (Petrarch Systems), a platinum-divinyltetramethyldisiloxane complex (3–3.5% platinum).
2.2 Sample Preparation
An addition cure reaction was employed to make the PDMS lenses. The bond formation is via a hydrosilylation reaction between the Si–H group of the cure agent and the C=C group of the divinyl-terminated PDMS in the presence of a platinum complex. The PDMS lenses were made by a procedure similar to that described by Chaudhury [35]. A glass slide was immersed in a hexane solution containing about 0.35% silane coupling agent ((tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane) for 1 min. The coupling agent reacts with the glass substrate via a hydrolysis reaction followed by a condensation reaction. The resulting surface has a low surface energy due to the fluorine groups. A melt that contained a divinyl-terminated PDMS, 0.2% of the platinum complex and 9–18% 182 cure agent was dropped on the pre-coated slide (using a pipette) to form hemispherical lenses. High-contact-angle drops were obtained due to the low surface energy of the pre-coated slides. The lenses were then transferred to an oven and cured at 65 oC for various amounts of time.
Some cured lenses were immersed in toluene for 1 h and then in a second bath of toluene for 1 h to remove free chains (sol fraction). In order to avoid cracking the samples, the swollen lenses were first partially deswollen in a 1:1 toluene–methanol mixture and then dried in air. A sol fraction of approximately 6% was determined by weighing the lens before and after the cleaning procedure.
In order to inhibit the occurrence of chemical reactions involving the terminal SiH group, a number of lenses were treated after cure with a thiol solution [0.5% 1-dodecanethiol, CH3(CH2)11SH in toluene] for 2 h. The sulfur groups poison the platinum catalyst contained in the lens thus inhibiting hydrosilylation between terminal silane groups and residual vinyl groups in the elastomer gel. The thiol treated lenses were subsequently washed in toluene for 1 h, deswollen in the 1:1 methanol–toluene for 1 h, and finally dried in air.
Polystyrene coated silicon substrates were made by spin coating (2000 rpm) a solution of 0.5% PS–Si–(O–CH2–CH3)3 in toluene onto a silicon wafer and then heating in a vacuum oven at 170oC for 1 h to complete the reaction of PS to the silicon substrate. The wafer with the grafted PS film was then washed with toluene to remove unreacted PS. The thickness of the grafted PS overlayer film was about 13 nm as measured by an ellipsometer.
The copolymer films were deposited onto the PS-coated substrates by spin coating a solution of the copolymer in toluene. The copolymer film thickness was varied by using different copolymer concentrations and spin rates. After spin coating, substrates were annealed in toluene vapor for 1 h and dried. The total film thickness, including the PS and copolymer layers, was measured by ellipsometry after annealing. The thickness of the copolymer film was then obtained by subtracting the thickness of the grafted PS layer from the total film thickness.
2.3 Measurements
A lens was placed with its curved face in contact with a coated substrate and then loaded with four glass slides (18 g). A thin transparent PS film was sandwiched between the slides and the lens to prevent the lens from sticking to slides after long loading times. After the load was removed, the contact radius between the lens and the substrate was measured as a function of time. After the contact radius measurement, the modulus and the radius of curvature of the same lens were measured. All the measurements were carried out at room temperature.
XPS measurements were performed with a Surface Science Instruments SSX-100 Model 05 small spot ESCA spectrometer, with a monochromatic Al Kα source. 1000 eV survey scans were performed using a 600-micron X-ray spot and 150-eV analyzer pass energy at a take-off angle θ of 60o, corresponding to a detection depth of approximately 7.5 nm. High-resolution spectra (pass energy = 50 eV) for C, O, and Si were taken at θ = 10o (detection depth about 1.5 nm) and θ = 60o using a narrow aperture (6o-acceptance slit).
An ellipsometer was used to determine the thicknesses of the PS and the block copolymer films. The instrument was a Rudolph Research AutoEl-II automatic ellipsometer with a laser light source of λ = 632.8 nm. The incident angle was 70o. A single layer program was used to calculate the film thickness from an input of the values of Δ and Ψ. The thickness on different spots of the film was measured to obtain an average value. A Rudolph in-situ 1100 microellipsometer with scanning stage was used to measure the small changes in thickness that occurred when the copolymer coated substrate was reacted with α,ω-divinyl-terminated PDMS. The latter experiment was employed to estimate the functionality of the copolymer brushes.
A Zeiss optical microscope with 10 × 6.3 lenses was used to obtain the contact radii of the PDMS lenses on the substrate and the radii of curvature of the lenses. The contact radius was measured using a reflection mode since the lens was on top of a silicon wafer. For the radius of curvature measurements, the lens was placed vertically against the wall of a transparent holder. A photograph of the lens was taken in a transmission mode. Finally the geometry of the hemisphere was analyzed to obtain the radius of curvature.
A load measuring device designed by Dr. C. Creton was used to obtain the moduli of the PDMS lenses. A PDMS lens (curved side down) on top of a PS substrate was placed on the device and loaded in the optical microscope. The contact radius of the lens on the substrate was determined as a function of applied force. The modulus of the lens was then calculated from the following equation [19]:
| (1) |
| (2) |
3 Results and Discussion
3.1 Organization and reactivity of the copolymer layers
When the P(S-b-DMS) diblock copolymer is spin-coated onto the PS coated substrate, it spontaneously self-organizes so that the PDMS blocks are adjacent to the air interface. This organization allows the PS blocks to interpenetrate with the grafted PS layer on the substrate and minimizes the surface energy of the system by locating the lower surface tension PDMS block at the air–polymer interface.
The as-coated copolymer layer was annealed in toluene vapor to facilitate the organization of both blocks to their preferred positions. The surface composition of the substrate coated with 15.0 nm of P(S-b-DMS)65 K was evaluated by X-ray Photoelectron Spectroscopy (XPS) before and after the toluene anneal. The atomic concentrations at detection depths of 1.5 nm and 7.5 nm are listed in Table 2. The Si/O ratio at low depths is close to unity, indicating that there is a layer of pure PDMS covering the substrate that is at least 1.5 nm thick. In the deeper probe, some polystyrene is detected and the Si/O ratio deviates from unity, indicating that the thickness of the pure PDMS surface is less than 7.5 nm thick. The lack of an annealing effect indicates that organization of the copolymer is sufficiently rapid to occur during the spin coating process. However, all samples were annealed in toluene vapor before use for consistency.
Atomic Compositions of P(S-b-DMS)65 K (15 nm film thickness)
| Conditions | Depth (nm) | %C | %O | %Si | O/Si | %PS |
| As deposited | 1.5 | 49.5 | 24.7 | 25.8 | 0.96 | 0 |
| 7.5 | 57.3 | 22.3 | 20.4 | 1.1 | 9 | |
| Annealed | 1.5 | 50.4 | 24.1 | 25.5 | 0.95 | 0 |
| 50.0 | 24.7 | 25.3 | 0.98 | 0 | ||
| 7.5 | 57.3 | 22.3 | 20.4 | 1.1 | 9 |
Contact angle measurements were carried out to characterize the surface composition of copolymer layers with various molecular weights. Fig. 1 shows contact angles of water for P(S-b-DMS)65 K layers as a function of the copolymer thickness at 30 oC. The first point corresponds to zero copolymer density, that is to say, pure PS. The measured value, 87.4o ± 0.6o, agrees well with the reported literature value of 87o for PS. [37] The contact angle increased sharply with the copolymer thickness and reached a constant value for thicknesses above 1.4 nm. The plateau value is about 104o, which is the same as the value for water on a PDMS monolayer [38]. This result implies that a layer of P(S-b-DMS)65 K with a thickness of 1.4 nm is thick enough to provide full coverage of PDMS over the PS substrate, consistent with what was found from XPS. The low value of contact angle at thickness of 0.8 nm may be due to the low coverage of the copolymer chains (perhaps forming patchy structures or mushrooms) on the PS substrate. A similar effect has been observed for the contact angles of water on a derivatized PMMA layer adsorbed on a gold substrate [39]. Water contact angles for copolymer layers with different molecular weights at fixed areal density are listed in Table 3. The contact angles are about the same for all the molecular weights, indicating that the surface is covered completely by PDMS at this chain density.
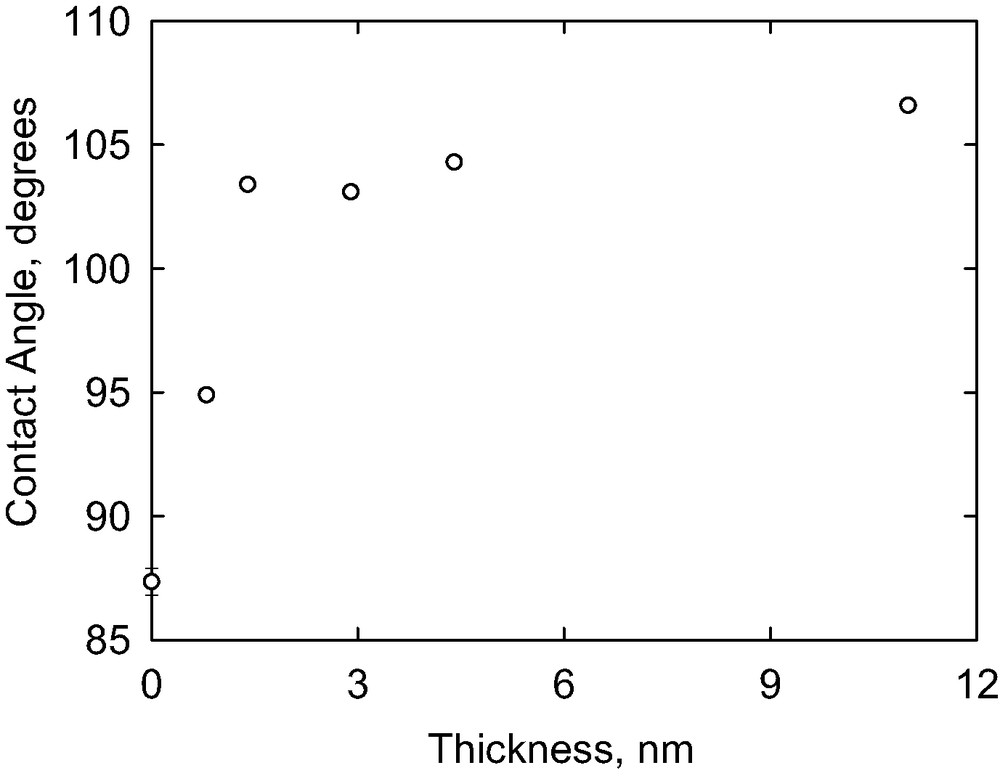
Water contact angle as a function of the 68 K copolymer overlayer thickness.
Water contact angles on P(S-b-DMS) coated substrates (areal density is 0.01 chains/nm2)
| Water contact angle (degrees) |
| P(S-b-DMS)47 K | 103 |
| P(S-b-DMS)54 K | 103 |
| P(S-b-DMS)60 K | 103 |
| P(S-b-DMS)65 K | 104 |
The reactivity of the Si–H groups of a copolymer layer were determined by measuring the amount of α,ω-vinyl-terminated PDMS (MW = 41 000) that could react with the layer. The measurement was done by first using the scanning ellipsometer to make a thickness scan across the sample then placing a catalyzed drop of the PDMS on the substrate for 24 h. The drop showed a finite contact angle on the substrate. After 24 h, the drop was washed off with hexane and a second thickness scan was made. As the ellipsometer showed a certain amount of long term drift it was essential to ensure that the drop covered only a fraction of the scan length.
Typical results concerning the change in ellipsometric angles Δ and Ψ between the two scans are shown in Fig. 2. The results in this figure correspond to a thickness change of 0.63 ± 0.04 nm. Studies of Langmuir–Blodgett films of functional PDMS showed that loop conformations with both chain ends attached to the water subphase were possible for molecular weights in excess of about 2000 Daltons [40]. Thus it is possible that either one or both ends of the divinyl PDMS may have reacted with the surface silane groups on the copolymer brush. Analysis of the thickness of the grafted PDMS layer leads therefore to estimates of the copolymer silane functionality in the range 7–14% for P(S-b-DMS)65 K. The low functionality may be the result of poor efficiency in terminating the living copolymer with dimethylchlorosilane or might be the result of a slow reaction of terminal silane groups with atmospheric moisture.

The change in ellipsometric angles Δ and Ψ caused by leaving on the substrate a drop of catalyzed vinyl-ended PDMS polymer for 24 h then washing it off. The drop was only on the right hand half of the scanned strip.
3.2 Elastic properties of the lenses
A load-and-unload experiment using the load measuring device was carried out to examine the mechanical hysteresis of the PDMS lenses. The applied force (P) was increased stepwise to the final load and then released stepwise to the initial load. At each load, the radius of the contact zone (a) between the lens and the PS substrate was measured. Fig. 3 shows the plot of a3 vs. P where the open circles and filled circles represent the loading and unloading cycles, respectively. Significant hysteresis was not observed. A similar test under condition of 16-h loading with 18 gm slides, again provided no evidence for hysteresis, illustrating that the lenses all exhibit linear elastic response.

Hysteresis test of the lens (i.e. contact radius cubed versus force) on a neat polystyrene substrate (circles: loading; squares: unloading).
3.3 Effect of experimental conditions on interfacial toughness
From a conceptual point of view, there are two mechanisms for an end-functional diblock copolymer to reinforce the interface between two immiscible homopolymers. If the rubbery copolymer sequence interpenetrates with the rubbery homopolymer gel, chain pull-out forces may lead to an increased interfacial toughness, as described by the weak junction theories of de Gennes and co-workers [20,21]. If the functional end group of the copolymer reacts with the gel, deformation of the interface becomes similar to the fracture of crosslinked rubbers, as considered by Lake and Thomas [27]. A number of experimental conditions were varied in order to determine the contributions of each of these possible mechanisms. In the case of an interfacial hydrosilylation reaction between the silane end group and residual vinyl groups in the PDMS gel, the degree of reaction and therefore the interfacial toughness was controlled by adjusting the reaction stoichiometry (i.e. the concentration of residual vinyl groups and copolymer end-groups) and by poisoning the platinum catalyst. When the catalyst is poisoned, the reaction does not occur and any increase in interfacial toughness noted can be ascribed to the effects of chain pull-out across the interface.
3.3.1 Effect of catalyst poisoning
A simple experiment was carried out to assess the importance of the hydrosilylation reaction that can occur across the interface. The cured lenses were separated into two batches. One batch of reference lenses was swollen with toluene to remove the sol fraction. The other batch was treated with a thiol solution as described earlier to deactivate the catalyst and hence inhibit the hydrosilylation reaction between the terminal Si–H groups on the PDMS blocks and residual vinyl groups of the gel. Adhesion experiments were carried out for both batches of lenses on PS–PDMS coated substrates and on substrates coated only with PS.
The results showed that the PS–PDMS layer increased the fracture toughness of the interface significantly for the reference lenses, for which the interfacial reaction could take place. For the thiol treated lenses, for which the reaction could not occur, the diblock copolymer had no effect on the interface adhesion and the interfacial toughness was about 0.05 J/m2, equivalent to the value obtained in the absence of copolymer.
The Dupre thermodynamic work of adhesion, Wa , between PS and PDMS can be calculated [37] from known interfacial energies:
| (3) |
3.3.2 Effect of the lens-curing conditions
The stoichiometry of the curing reaction influences the adhesion measurement in two ways. First, the concentration of hydromethyl siloxane curing reagent used controls the molecular weight between crosslinks and therefore the modulus of the gel. Second, the concentration of residual vinyl groups available for the interfacial hydrosilylation reaction is related to the concentration of excess vinyl groups in the gel formulation. Fig. 4 shows the effect of cure agent concentration on the contact radius. As the cure agent concentration increases, the interfacial toughness (i.e. G ∝ a3, see equation (2)) decreases steadily, reflecting the associated decrease in residual vinyl groups. The decrease in toughness continues beyond the stoichiometric concentration of 15%, an expected result since it is likely that the curing reactions at the surface do not go to completion at stoichiometric conditions (Fig. 5).
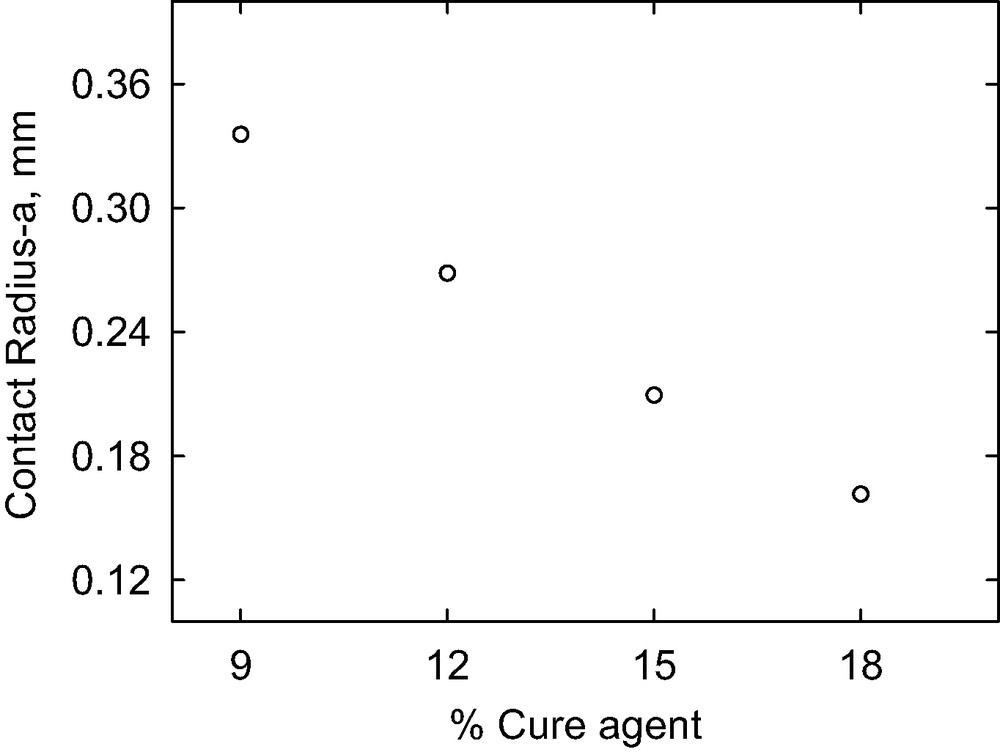
The effect of cure agent concentration on contact radius.
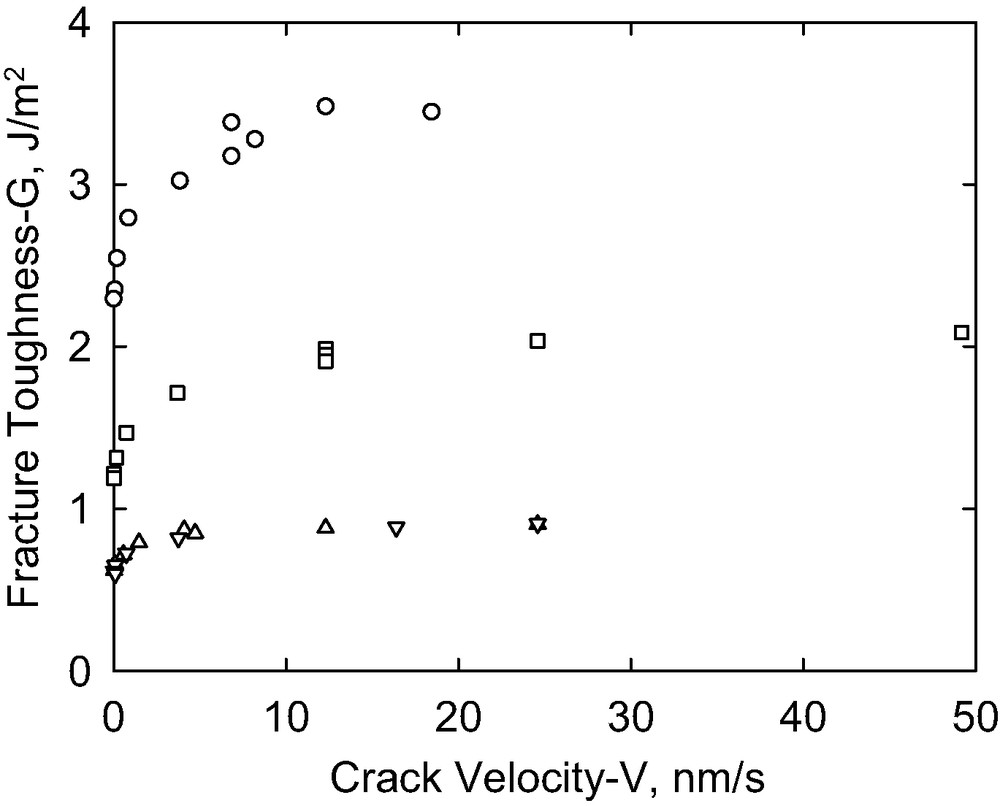
The fracture toughness as a function of the crack propagation rate for different cure times (circles: 15 min; squares: 30 min; up triangles: 60 min; down triangles: 90 min).
The effects of the gel stoichiometry upon adhesion were also studied by comparing contact radii of equivalent gels on P(S-b-PDMS) coated substrates with contact radii on PS substrates without copolymer coatings. The copolymer layers enhanced the adhesion only for concentrations of cure agent less than the approximately stoichiometric value of 15%, that is, when there are residual vinyl groups available to react with the silane end groups in the copolymer layer. At a cure temperature of 65 °C, the contact radii on both substrates were independent of cure time for times of 60 min and longer.
Fig. 6 demonstrates that the interfacial fracture toughness is also dependent upon cure time. The curing reaction involves reaction of the vinyl groups of the divinyl-terminated PDMS with the Si–H groups of the cure agent in the presence of a platinum catalyst to form the cross-linked lenses. As the cure reaction proceeds, progressively fewer residual vinyl groups tethered to the gel are available to participate in the interfacial reaction with the silane functional block copolymer layer with which it is in contact. Two regions of behavior are evident in the figure: a low rate region where the toughness increases rapidly with the crack growth rate, and a high-rate region where the toughness reaches a saturation value and increases only slightly with crack rate. The dependence of G upon V is approximately linear in the slow-rate region, allowing for determination of the threshold toughness, Go, by extrapolation to zero crack rate. The threshold toughness (Fig. 6), decreases with increasing the cure time.
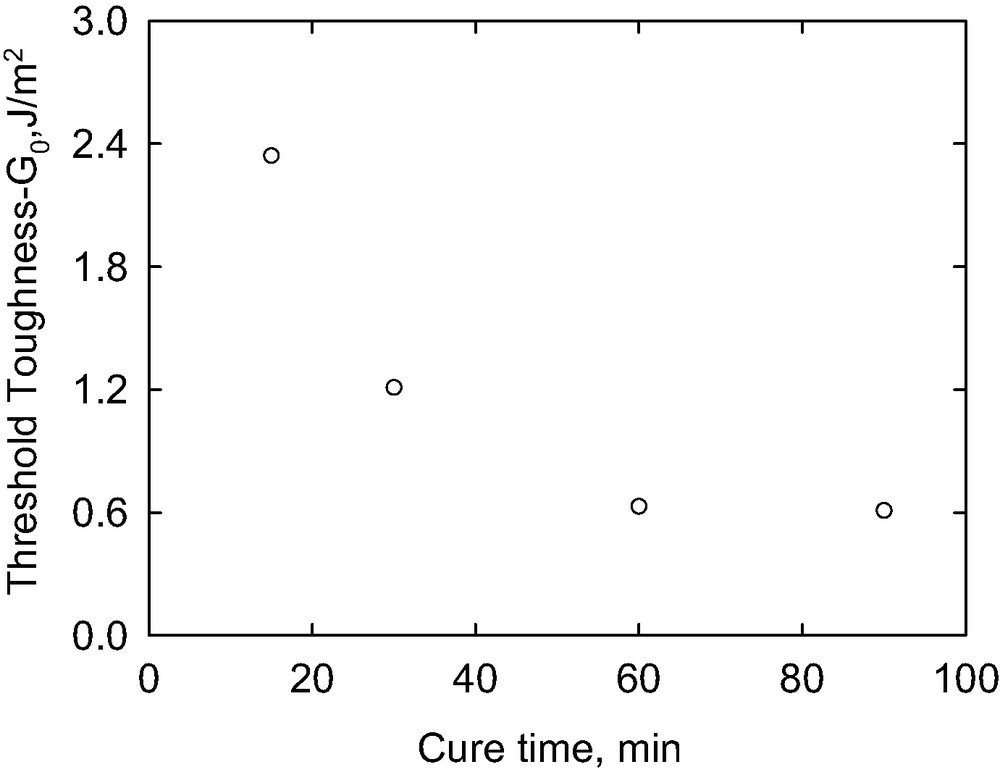
Extrapolated threshold toughness as a function of the cure time.
It is evident from these results that the adhesion enhancement afforded by the reactive end-functional block copolymer originates from the interfacial hydrosilylation reaction between the silane groups on the copolymer chain end and unreacted vinyl groups in the PDMS lens. A standard procedure for fabricating lenses was therefore adopted for further experiments aimed at defining more quantitatively the effects of chemical bridging across an interface on the interfacial fracture toughness. The concentration of cure agent had to be low enough to leave enough free vinyl groups to react with the silane end groups of the copolymer, yet large enough to produce a reasonable modulus in the lens. Thin copolymer layers were used to provide a low areal density of silane end groups so that residual vinyl groups from the gel could be considered to be in excess. The silane groups were therefore the limiting reactant for the interfacial hydrosilylation reaction. The curing time was chosen to be long enough to ensure complete reaction of the crosslinking agent (i.e. silane was the limiting reactant for the elastomer gel) before the lens was placed in contact with the substrate.
The standard procedure adopted was to use 9% cure agent, to cure at 65 oC for 2 h, to wash in toluene and to dry as described in the experimental section. Removing unreacted divinyl PDMS chains ensured that all residual vinyl groups were tethered to the gel such that further reaction with the silane end group of the copolymer chains resulted in a covalent bridge across the interface. The effective modulus K of the standard lenses obtained from the average value of 25 samples was 2.28 ± 0.15 MPa. The average molecular weight between crosslinks estimated from the affine model [41] was 5700 g/mole. The lenses have approximately 60% excess of vinyl groups based upon the stoichiometry and reported functionality of the lens precursor polymers.
3.3.3 Effect of dwell time
In the adhesion measurements, the lens was forced into contact with the copolymer coated substrate by applying a weight for a fixed dwell time, after which the weight was removed. Fig. 7 illustrates the effect of dwell time for standard lenses loaded on P(S-b-DMS)65 K substrates (2.4-nm copolymer thickness). At a dwell time of four hours, the copolymer had no influence on the interfacial toughness. The lens detached from the substrate instantaneously when the load was released, similar to the behavior observed for the neat PS/PDMS interface. When the dwell time was increased to 8 h, the copolymer started to show enhanced adhesion. A slight time dependence of G was observed in the slow rate region and the value of G0 was three times the work of adhesion (Wa) for a neat PS/PDMS interface. The threshold fracture toughness, shown in Fig. 8, achieved its maximum value for a dwell time of about 16 h. In this case, the value of G0 was about eight times that of Wa. Dwell times longer than 16 h did not change the fracture toughness significantly.

The fracture toughness as a function of the crack propagation rate for different dwell times (dashes: 4 h; circles: 8 h; squares: 16 h; triangles: 24 h).
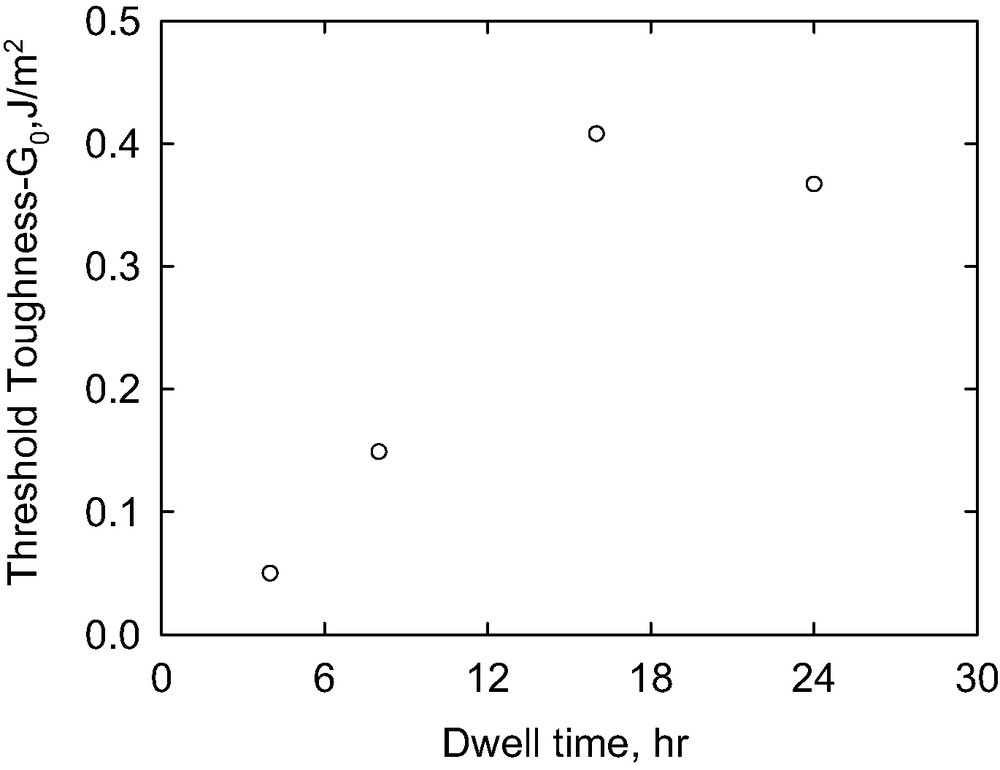
Extrapolated threshold toughness as a function of the dwell time.
In the case of copolymer-modified interfaces, the observed increase in interfacial toughness with dwell time [42,43] has been attributed to a number of mechanisms. An elastomer, which undergoes deformation, will creep and wet the surface. This process is found to be particularly important when the materials have rough surfaces [44,45]. Brown and co-workers [13,14] found that, for a polystyrene (PS)/polyisoprene (PI) system, the equilibrium dwell time for the chain pull-out experiment was about 4 h, while that for the peel test was about 10 days. The difference was attributed to the difficulty in obtaining smooth and clean surfaces to be joined for the peel test, so the long process was connected with slow surface flattening. The second mechanism is the diffusion of the free ends of the polymeric chains across the interface [46–49]. Dwell time effects can also be caused by surface blooming where vulcanizing ingredients from the bulk of rubber migrate to the surface. This process increases the surface energy of the materials and subsequently their adherence.
It is unlikely that the dwell time effect in the present system was due to any of these previously reported effects. The lenses were cleaned with toluene directly before loading so that surface bloom effects can be ruled out. The lack of interfacial reinforcement when the catalyst is poisoned also tends to rule out any time effects due to the kinetics of chain interpenetration. Furthermore, the dwell times employed herein are significantly shorter than those found to be necessary for the interpenetration of tethered PDMS brushes into PDMS gels. [25]. The dwell time effect in our study is therefore most likely associated with the kinetics of the interfacial hydrosilylation reaction between the terminal silane groups of the PDMS blocks and the vinyl-terminated PDMS dangling chains in the elastomer network. Since a loading time of 16 h was found to produce an apparent steady state toughness for the longest block copolymer, we have fixed the dwell time at 16 hours in the remainder of the experiments.
3.3.4 Effect of copolymer areal density
Fig. 9 shows the variation of toughness with the crack propagation rate for a range of copolymer areal densities (with a copolymer layer thickness up to 2.9 nm) for P(S-b-DMS)65 K. The toughness was increased by a factor of 16 by the presence of only 0.016 chains/nm2 at the interface. Two regions of behavior are again evident in these data. Fig. 10 illustrates that the dependence of G on V is approximately linear in the slow rate region allowing determination of the threshold toughness, Go, by extrapolation to zero crack velocity.

The fracture toughness as a function of the crack propagation rate for different areal density (chains/nm2) of the copolymer chains. (circles: 0.004; squares: 0.008; up triangles: 0.012; down triangles: 0.013; diamonds: 0.016) [P(S-b-DMS)65 K].
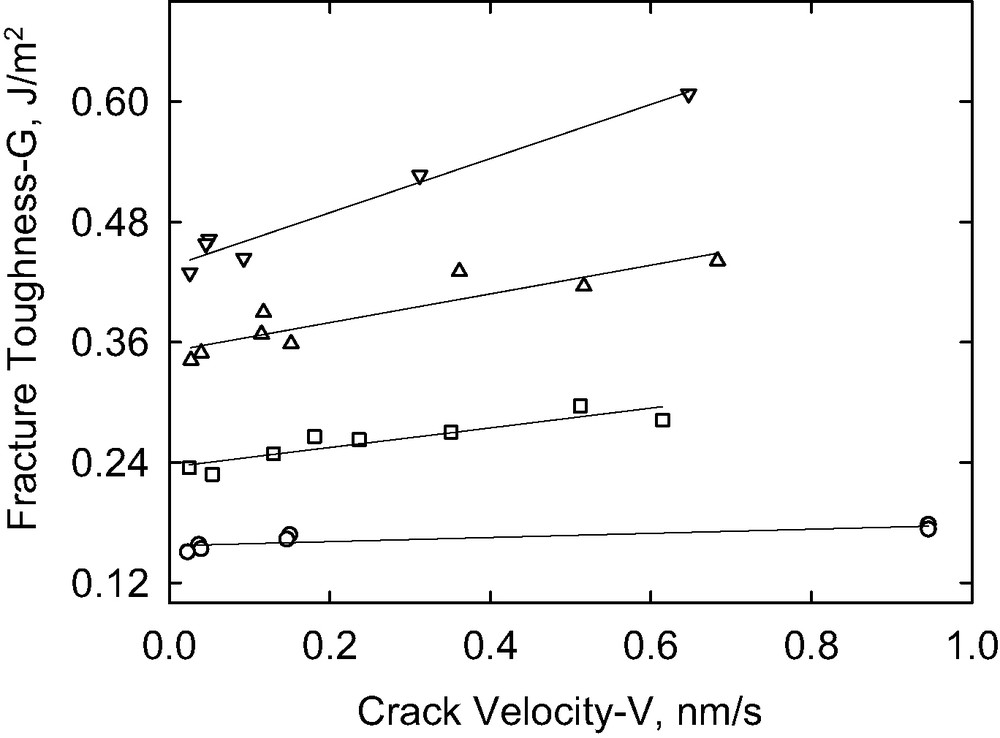
The fracture toughness as a function of the crack propagation rate in the slow rate linear regime for different areal density (chains/nm2) of the copolymer chains (circles: 0.004; squares: 0.008; up triangles: 0.012; down triangles: 0.016) [P(S-b-DMS)65 K].
It is noteworthy that crack velocities less than 1 nm/s are required to reach behavior that can be successfully extrapolated to zero crack velocity. Similar qualitative behavior was observed previously for interfaces toughened with P(S-b-I) diblock copolymers [13,14]. A rapid linear increase in G for velocities above 2 nm/s was attributed to energy losses associated with pullout of bridging copolymer chains from the bulk elastomer and a slight increase of G with crack velocity in the high rate region was attributed to bulk viscous losses in the lens. The linear dependence at low rates is coincident with the prediction from chain pullout models [50]. It is unlikely, however, that these same interpretations can be applied to the present case, where the adhesion enhancement originates from the interfacial reaction between the diblock copolymer and the network.
The threshold toughness, at low concentrations, depends linearly on the areal chain density of the copolymer P(S-b-DMS)65 K, Σ, as shown in Fig. 11. This result is consistent with an adhesion mechanism involving the formation of covalent bridges between the copolymer and gel. If the degree of reaction of the copolymer silane end groups is independent of the areal density, it is apparent that the density of covalent bridges across the interface is directly proportional to the copolymer areal density. When a crack propagates along the interface, the copolymer chains that have reacted at the interface must either break or pull out from the PS under layer. The possibility of chain pullout in a glassy polymer such as PS is controlled by the molecular weight of the polymer. When the molecular weight increases above about 1 to 3 times the entanglement molecular weight, Me, the pullout becomes sufficiently difficult that failure occurs by chain scission [51,52]. The longer two diblock copolymers used in this work had a molecular weight of about 2.2 Me, while the shorter pair had a PS molecular weight of about 1.4 Me. It would seem likely that failure occurred by chain scission for the longer copolymers, but pullout of the PS block from the PS substrate is possible for the shorter diblock copolymers.
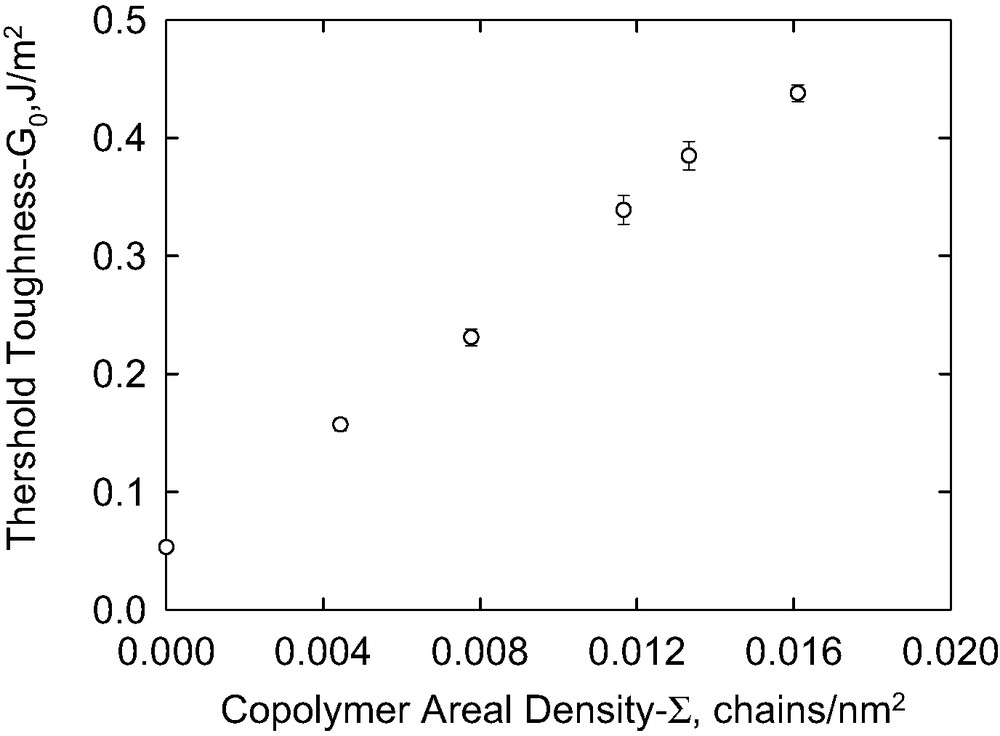
Extrapolated threshold toughness as a function of block copolymer areal density [P(S-b-DMS)65 K].
The threshold toughness for P(S-b-DMS)60 K attains a constant value at areal densities above an apparent surface saturation concentration as shown in Fig. 12. Surface saturation occurs when the copolymer layer reaches complete monolayer coverage. At higher concentrations, the formation of islands can be detected visually, indicating the formation of multilayers. Once multilayers are formed, the interfacial bridging mechanism is compromised. Thus one expects a maximum adhesion enhancement for a concentration corresponding to one complete monolayer for which the surface density of reactive silane groups is optimal.
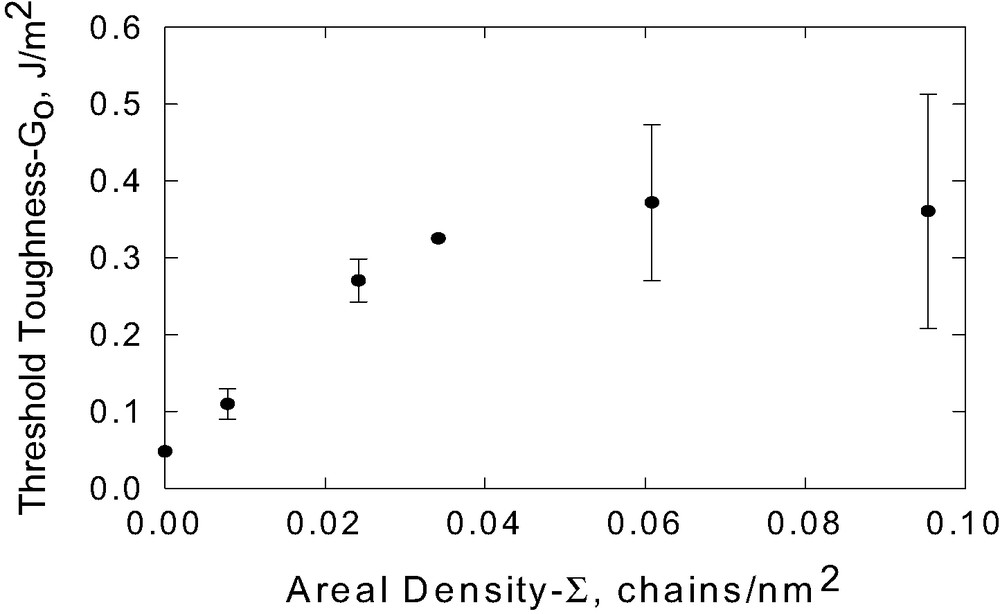
Extrapolated threshold toughness as a function of block copolymer areal density [P(S-b-DMS)60 K].
The interfacial toughness measurements provide strong evidence that the adhesion enhancement for our interfaces modified with reactive end-functional block copolymers is the result of covalent bridges formed across the interface between the copolymer brush and the elastomer gel. This situation is therefore similar to the failure of an elastomer network, for which bridging chains must undergo scission. Lake and Thomas [27] proposed a model for failure of crosslinked elastomers that considers the energy required to break a bridge or strand, defined as the length of chain between cross-links. The strand is stretched until each main chain bond has a stretching energy equal to its dissociation energy, U, and then ruptures, releasing the stored energy as heat. If there are n main chain bonds per repeat unit, Nx repeat units between crosslink points and Wa is the work of adhesion, the theory predicts
| (4) |
The model has been shown to work well for low speed, high temperature cohesive failure of a number of well-crosslinked elastomers. However in cohesive failure of a network Σ and Nx are not independent variables but combine to form a term dependent on Nx1/2 so the model cannot be fully tested. The situation where the degree of polymerization between crosslinks is low, Nx > Ne is not as well studied as the well crosslinked situation when Nx < Ne. Mazich et al. [53] showed that when Nx > 2 Ne, Go no longer increased as Nx1/2, but was approximately independent of Nx.
It is valuable initially to compare the data in Fig. 11 with the prediction of the Lake–Thomas model, ignoring any entanglement issues. From the gradient of this plot we obtain a fracture energy of about 2.4 × 10−17 J/chain. However earlier model experiments showed that only 7–14% of the copolymer chains react so the an apparent fracture energy per reacted chain is estimated to be 1.7–3.4 × 10−16 J. The dissociation energy for the Si–O bonds of the PDMS copolymer sequence is 8.9 × 10−19 J, while that of the C–C bonds in the polystyrene backbone and the Si–C linkages connecting the PDMS sequence to both the gel and the styrene sequence is 6.1 × 10−19 J. Fracture is therefore expected to occur at the gel–copolymer junction or within the polystyrene sequence when the stored energy for each bond in the PDMS strand reaches 6.1 × 10−19 J. Using this value, the fracture energy is consistent with a number of 280–560 main chain bonds, or a molecular weight of 10 000–20 000 in the fractured strand. This number has to be compared with the actual molecular weight of the strand that couples the network to the substrate. The PDMS part of the copolymer had a GPC average molecular weight of 65 000 and it is assumed to end react with a network strand of molecular weight of about 6 000 for a total strand molecular weight of 74 000. Clearly the actual molecular weight is several times larger that calculated from this direct application of the Lake–Thomas model.
At this point it is valuable to consider how well the Lake–Thomas model works for well-crosslinked PDMS networks where entanglement is not an issue. The Lake–Thomas theory for cohesive failure in a well crosslinked network can be applied to calculate an effective value of 5.0 × 10−19 J for the bond-dissociation energy from the data of Gent and Tobias [54] and of Yanyo and Kelley [55] quoted by Mazich et al. Clearly the Lake–Thomas theory works very well. The most likely explanation for the result obtained here is that, when the length of the strand between crosslink points is much greater than the entanglement length, the energy loss corresponds to just a length controlled by the entanglement spacing (the molecular weight between entanglements is about 10 000 for PDMS [56]). Similar criteria have been proposed previously for cohesive failure but in cohesive failure it is not possible to separate the number of strands per unit area from their effective length. The results presented here suggest that the effective length for energy loss is one to two times the entanglement spacing.
3.3.5 The effect of copolymer molecular weight
The effects of molecular weight are illustrated in Fig. 13, which shows the variation of fracture toughness with the crack growth rate for 4 copolymers of differing molecular weight but fixed areal density (0.013 chains/nm2). The two lower molecular weight copolymers provided only a small enhancement in toughness compared to the bare PS substrate. These copolymers had relatively short PS blocks and perhaps failure occurred by pullout from the PS underlayer. P(S-b-DMS)65 K gave a significantly higher toughness than P(S-b-DMS)60 K. The reason for this result is not clear as, for these copolymers, the PS molecular weights were the same, PDMS molecular weight difference was small and the reactivity of the Si–H groups was about the same. The threshold toughness obtained by extrapolation of the low rate data (see Fig. 14) does increase with molecular weight as shown in Fig. 15. The errors in the data however are too large however to determine whether the threshold toughness scales linearly with molecular weight of the PDMS copolymer sequence as would be predicted by the Lake–Thomas theory.

The fracture toughness as a function of the crack propagation rate for different molecular weights of the PDMS block all at Σ ≈ 0.017 chains/nm2. The molecular weights of the block copolymers in kDaltons are: 47 (circles), 54 (squares), 60 (up-triangles) and 65 (down-triangles).
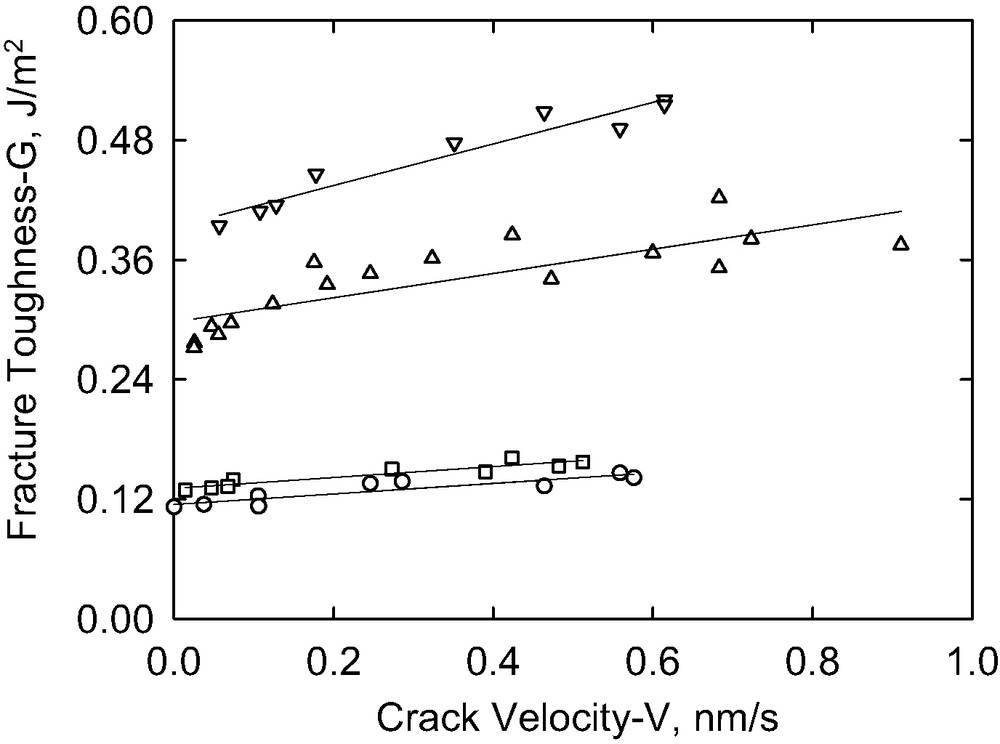
The fracture toughness as a function of the crack propagation rate in the slow rate linear regime for different molecular weights of the PDMS block all at Σ ≈ 0.017 chains/nm2. The molecular weights of the block copolymers in kDaltons are: 47 (circles), 54 (squares), 60 (up-triangles) and 65 (down-triangles).
Extrapolated threshold toughness as a function of the PDMS block molecular weight.
The data also suggest the existence of a threshold PDMS block molecular weight of about 55 000 that is not predicted by the theory. That is, an enhancement in threshold toughness over that of the bare substrate is observed only for PDMS sequence molecular weights above an apparent threshold of about 55 000. The results suggest that the length of PDMS chain that contributes to the fracture toughness is much shorter than the entire sequence length. This conclusion is supported by our previous analysis of the fracture energy per strand that indicated a strand length of only 13–26% of the total sequence length.
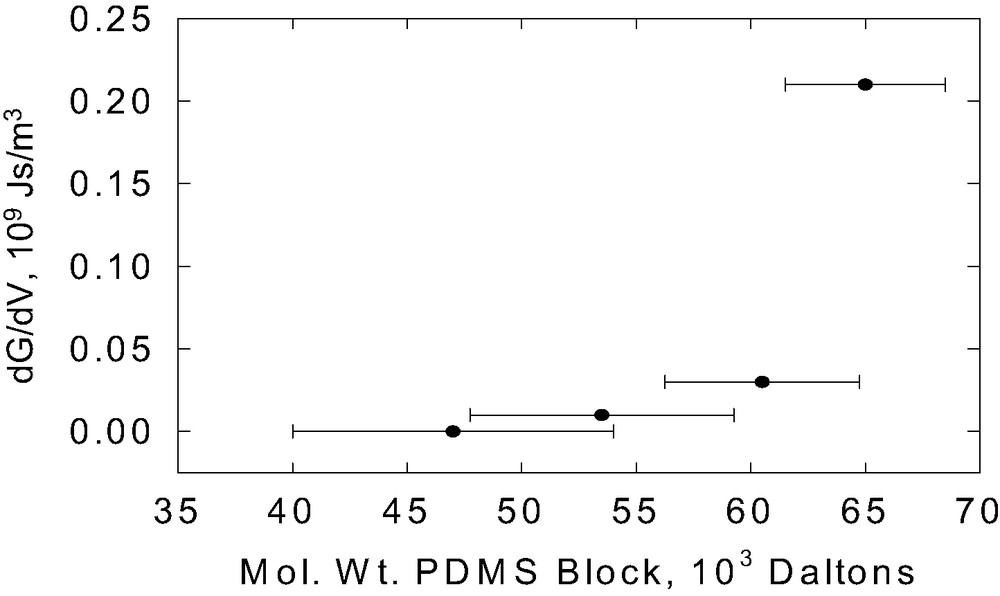
The energy dissipation model proposed here to explain Go, which is based on the Lake–Thomas model, does not require that the PDMS chains from the copolymer penetrate into the network, it only requires that they react with the network. However, as is evident from Figs. 7,9 and 13, we also see considerable rate dependence in G at low crack velocities. The rate dependence is linear in crack velocity, scales linearly with areal density of the block copolymer (see Fig. 16) and increases with molecular weight (see Fig. 17). Hui et al. [57] have very recently proposed a model for the crack growth rate dependence of fracture that occurs by chain scission based on a previous model by Chaudhury [58,59]. They predict that, at very low crack velocities, G varies linearly with V, but this regime only occurs at velocities so low that (G – W)/W <<1. At high velocities they predict that G ~ (ln V)2. We do not have data over a sufficient range of V to compare with this prediction.
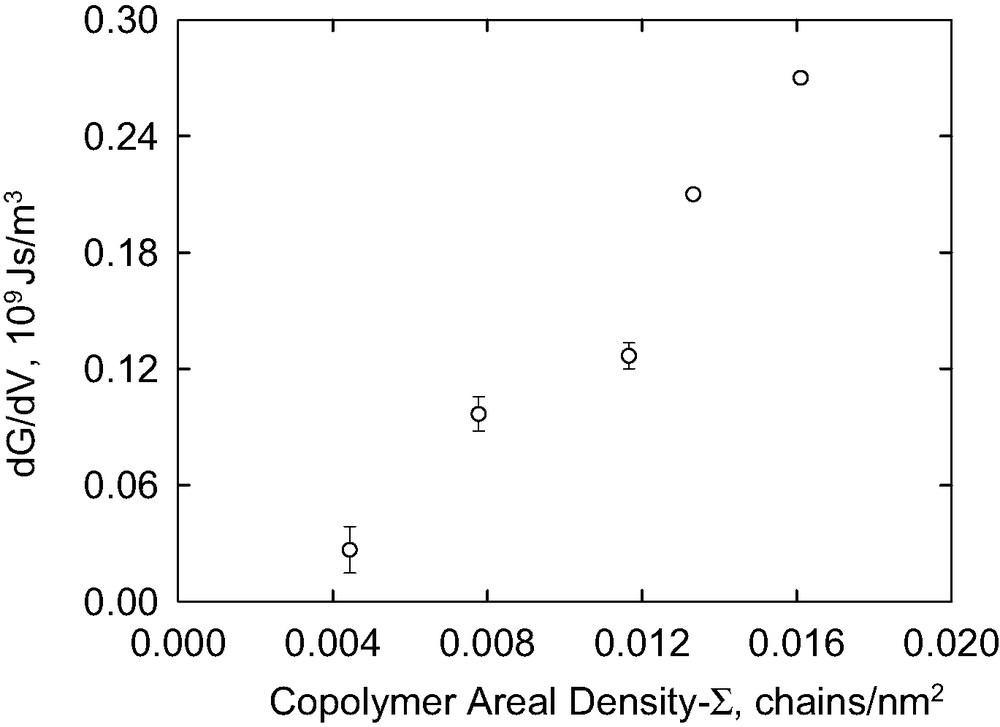
The slope of low rate data, dG/dV, as a function of block copolymer areal density.
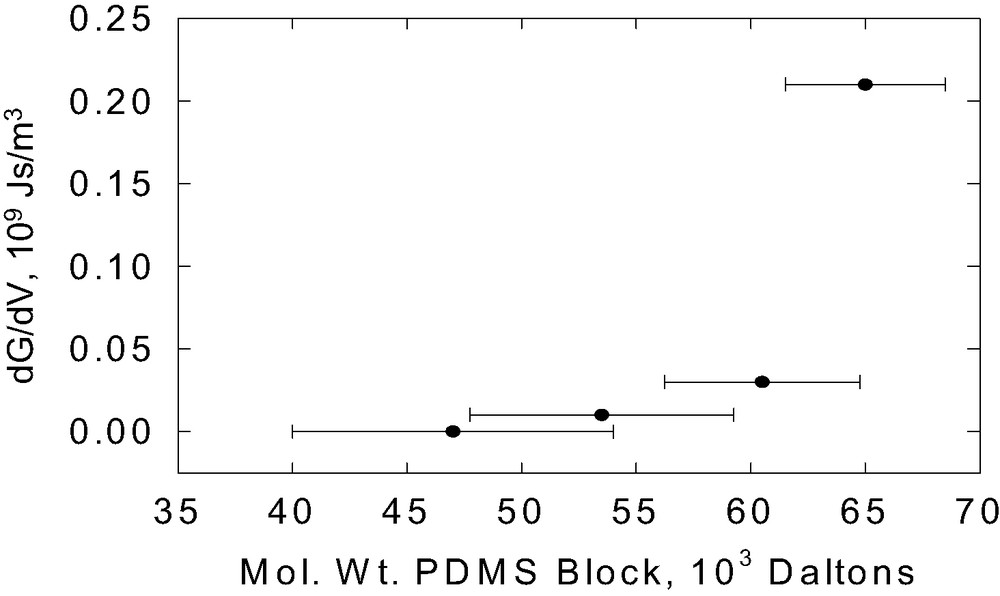
The slope of low rate data, dG/dV, as a function of PDMS block molecular weight.
4 Conclusions
The fracture toughness of the interface between a PS substrate and a PDMS elastomeric gel was increased by placing a thin layer of silane end-functional diblock copolymer at the interface. The threshold toughness reduced to the pure thermodynamic work of adhesion between PS and PDMS homopolymers in the absence of block copolymer. The magnitude of adhesion enhancement was found to be proportional to the concentration of residual vinyl groups in the PDMS elastomer and vanished when the platinum catalyst was poisoned, indicating that adhesion enhancement originates from the interfacial hydrosilylation reaction between terminal copolymer silane end groups and residual vinyl functionality on dangling chains in the PDMS elastomeric network. At low rates, the fracture toughness increased linearly with the crack propagation rate, and gradually approached a saturation value. At a fixed rate, the fracture toughness increased linearly with the areal density of the copolymer. A toughness increase factor of 16 was afforded by the presence of 0.016 chains/nm2 copolymer molecules at the interface. The threshold toughness, obtained by extrapolation to zero crack velocity, increased linearly with the copolymer areal density up to a concentration corresponding to full monolayer coverage. When the amount of copolymer at the interface was fixed, the toughness increased with increasing molecular weight of the PDMS block. These results are compared to the predictions of the Lake-Thomas model for fracture of elastomers.
Acknowledgments
This material is based upon work, by the University of Connecticut researchers, supported by, or in part by, the U.S. Army Research office and grant number DAAH04-95-1-0592. The University of Connecticut authors also wish to acknowledge funding provided by the Polymer Blend Compatibilization Consortium and the 3 M company. The interaction with the Strasbourg group was funded by a joint NSF–CNRS collaborative research grant. The authors are grateful to several research colleagues from the IBM Almaden Research Center in San Jose for providing their expertise and services: Dr. D. Miller for carrying out the XPS measurements, Dr. P. Cotts for characterization the molecular weights of the copolymers and Dr. K. Shull for synthesizing the silane-functional polystyrene.