

1 Introduction
La chimie des complexes de métaux de transition a fait l'objet de nombreux travaux au cours de ces dernières années [1–5]. Elle a connu un développement spectaculaire du fait de ses nombreuses applications en catalyse, de l'étude des sites actifs des métalloprotéines et de la perspective de l'émergence de composants moléculaires adaptés au traitement de l'information [6].
Dans ce travail, nous présentons la synthèse de ligands tridentates (H2LA, H2LB et H2LC) et de leurs complexes avec des sels de cuivre(II) et nickel(II). Les composés obtenus sont caractérisés par différentes techniques spectroscopiques, IR, UV, et RMN pour les ligands. Nous avons aussi étudié les interactions métalliques à travers les mesures de susceptibilités magnétiques et la voltamétrie cyclique.
2 Partie expérimentale
L'hydrazide de l'acide acétique, le 3-méthoxy salicylaldehyde, le salicylaldéhyde et le 2-aminophénol sont d'origines Janssen et Aldrich. Les sels métalliques sont des produits Prolabo, Riedel, Labosi et Janssen. Les solvants sont d'origine Aldrich. Tous ces produits, d'origine commerciale, ont été utilisés sans purification préalable.
Les analyses élémentaires reportées dans le Tableau 1 ont été effectuées à l'aide d'un instrument Carlo Erba CHNS-OEA 1108 « Elemental Analyser » au service de microanalyse (Dipartimento di chimica inorganica metallorganica ed analitica) de l'université de Padoue (Italie).
Analyse élémentaire, point de fusion, rendement et couleur des composés H2LA, H2LB, H2LC, A, B, C, D et E
| Réf. | F.B. | Trouvé (calculé) | P.F. (°C) | R∗ % | Couleur | ||
| %C | %H | %N | |||||
| H2LA | C9H10N2O2 | 59,41 (60,65) | 5,61 (5,65) | 15,65 (15,72) | 199 | 89 | Blanc |
| H2LB | C10H12N2O3 | 56,88 (57,68) | 5,71 (5,81) | 13,38 (13,45) | 175 | 83,2 | Blanc |
| H2LC | C14H13NO3·1/4H2O | 67,22 (67,87) | 5,14 (5,49) | 5,72 (5,65) | 189 | 89 | Rouge |
| A | CuC10H12N2O3(NO3)2·EtOH | 31,80 (32,62) | 3,35 (4,11) | 12,27 (12,68) | >250 | – | Vert foncé |
| B | CuC10H12N2O3Cl2·3/2H2O | 32,89 (32,49) | 3,85 (4,09) | 7,74 (7,57) | 182 | 54,1 | Vert olive |
| C | CuC10H12N2O3Br2 | 28,64 (27,83) | 2,96 (2,80) | 6,82 (6,49) | 185 | 57,5 | Vert olive |
| D | CuC14H11NO3 | 54,87 (55,17) | 3,56 (3,64) | 4,70 (4,59) | >250 | 66,66 | Vert olive |
| E | NiC9H8N2O2·3/2H2O | 41,05 (41,23) | 4,21 (4,20) | 10,47 (10,69) | >250 | 95,5 | Orange |
Les spectres infrarouge ont été enregistrés en suspension dans le nujol à l'aide d'un spectromètre PerkinElmer 580 (4000–400 cm−1) – les faces utilisées sont en bromure de potassium – et d'un spectromètre FTIR Nicolet pour les basses fréquences (600–50 cm−1).
Les points de fusion sont déterminés avec un appareil Buchi 530, en utilisant des tubes capillaires.
Les spectres RMN ont été enregistrés, en solution dans le CD3CN, à l'université de Rouen (France) à l'aide d'un spectromètre Bruker ARX 400 MHz.
Les spectres UV en solution ont été enregistrés par un spectromètre UV de type JASCO modèle 7800 à double faisceau. Les cellules sont en quartz et le solvant utilisé est l'acétonitrile. Les spectres UV solides ont été enregistrés à l'université de Florence (Italie) avec un spectromètre Cary5 UV.Vis NiR.
Les mesures de susceptibilité magnétique à température variable sont effectuées à l'aide d'un appareil Metronique Ingegnerie SQUID M03 opérant entre 0 et 8 T dans la région 2–300 K. Le composé est maintenu sous atmosphère d'hélium. Les données magnétiques sont corrigées de la contribution diamagnétique, estimée à partir des constantes de Pascal.
Les mesures électrochimiques ont été réalisées à l'aide d'un potentiostat modèle 362 équipé d'une table traçante Sefram. Les mesures voltampérométriques ont été effectuées dans une cellule équipée de deux électrodes de platine et d'une électrode Ag/AgCl comme référence. Le complexe étudié est dissous dans de l'acétonitrile contenant du tétrabutyl ammonium perchlorate (0,1 M) comme électrolyte support.
2.1 Synthèse des ligands
2.1.1 3-Méthoxysalicylaldéhyde acétylhydrazone C10H12N2O3 (H2LA)
On a introduit, dans un ballon de 250 ml, 3,01 g (40,67 mmol) d'acétylhydrazine, auxquels on a ajouté, goutte à goutte, sous agitation et sous un léger chauffage à 50–60 °C, 60 ml d'une solution du 3-méthoxysalicylaldéhyde (6,15 g ; 40,46 mmol) dans un mélange H2O/EtOH (50:50). Le mélange a été maintenu dans ces conditions pendant 4 h. Après refroidissement, nous avons obtenu un précipité blanc, qui a été récupéré par filtration, lavé avec un mélange (H2O/EtOH), puis à l'éthanol absolu refroidi. Il a été séché dans un dessiccateur sous P4O10, après recristallisation dans du chloroforme.
RMN 1H (δH, CD3CN) ; 11,01 (1H, s, O–H) ; 8,25 (1H, s, HCN) ; 8,10 (1H, s, N–H) ; 6,80–7,11 (3H, m, Ar) ; 3,90 (3H, s, H3C–O) ; 2 (3H, s, H3C–CO).
RMN 13C (δC, CD3CN) : 166,8(CO) ; 149,3(CAr–OMe) ; 149,2(CN) ; 149(CAr–OH) ; 123,5(CAr) ; 122,5(CAr) ; 120,5(CAr) ; 114,7(CAr) ; 56,8(CH3–O) ; 21,6(CH3[Ac]).
2.1.2 3-Méthoxy-(2′hydroxyphényl)salicylaldimine C14H13NO3·1/4H2O (H2LB)
4,56 g (30 mmol) de 3-méthoxysalicylaldéhyde sont dissous dans 50 ml d'éthanol absolu. À ce mélange ont été ajoutés goutte à goutte 15 ml d'une solution éthanolique contenant 3,27 g (30 mmol) de 2-aminophénol. Le mélange a été chauffé à reflux et maintenu sous agitation durant 2 h. La solution a ensuite été refroidie dans un bain de glace. Le produit rouge obtenu est lavé à l'éthanol refroidi, puis à l'éther. Le produit, recristallisé dans du méthanol, a ensuite été séché sous P4O10 au dessiccateur.
RMN 1H (δH, CD3CN) : 13,3 (2H, s, O–H) ; 8,8 (1H, s, HCN) ; 6,8–7,3 (7H, m, Ar) ; 3,90 (3H, s,H3C–O).
RMN 13C(δC, CD3CN) : 164,6(CN) ; 152,3(CAr–OH) ; 151,4(CAr–OMe) ; 149,5(CAr–OH) ; 136,9(CAr–N) ; 129,3(CAr) ; 125,1(CAr) ; 121,7(CAr) ; 119,9(CAr) ; 119,6(CAr) ; 117,2(CAr) ; 116,5(CAr) ; 56,8(CH3–O).
2.1.3 Salicylaldéhyde acétylhydrazone C9H10N2O2 (H2LC)
On a introduit, dans un ballon de 250 ml, 6 g (81 mmol) d'acétylhydrazide, auxquels on a ajouté 60 ml d'un mélange eau/éthanol (50:50). À cette solution, maintenue sous agitation et sous chauffage léger (50 à 60 °C), on a ajouté goutte à goutte 9,892 g (81 mmol) de salicylaldéhyde dans 40 ml de mélange eau/éthanol (50:50). Le mélange obtenu a été maintenu à reflux pendant 2 h. Un précipité blanc est apparu, récupéré par filtration, lavé avec le même solvant (H2O/EtOH), et ensuite à l'éthanol refroidi. Il a été séché sous P4O10 au dessiccateur, après recristallisation dans du chloroforme.
RMN 1H (δH, CD3CN) : 11,4 (1H, s, O–H) ; 8,2 (1H, s, HCN) ; 8,10 (1H, s, N–H) ; 6,9–7,3 (4H, m, Ar) ; 2,1 (3H, s, H3C–CO).
RMN13C (δC, CD3CN) : 166,8(CO) ; 159,3(CAr–OH) ; 149,3(CN) ; 146,9(CAr–CN) ; 132,4(CAr) ; 132(CAr) ; 120,8(CAr) ; 120,4(CAr) ; 21,6(CH3–[Ac]).

2.2 Synthèse des complexes
2.2.1 {Cu(H2LA)NO3}(NO3)2EtOH [A]
À 30 ml d'une solution d'éthanol à chaud contenant 422 mg (2 mmol) de H2LA ont été ajoutés 494 mg (2,04 mmol) de nitrate de cuivre(II) trihydraté Cu(NO3)2·3H2O dissous dans 5 ml d'éthanol. Le mélange a été chauffé sans réfrigérant, tout en maintenant l'agitation afin de réduire le volume. La solution obtenue a été laissée au repos durant deux jours. Les cristaux ont ensuite été récupérés par filtration, lavés à l'éthanol puis à l'éther diéthylique, enfin séchés sous P4O10.
2.2.2 {Cu(H2LA)Cl2}2(H2O)3 [B]
422 mg (2 mmol) de H2LA ont été introduits dans 30 ml d'éthanol absolu. La solution a été chauffée légèrement jusqu'à dissolution totale ; on a ensuite ajouté 10 ml d'éthanol, contenant 341 mg (2 mmol) de CuCl2·2H2O et 1 ml de HCl 2 M. Le mélange a été porté à reflux pendant 20 min et maintenu sous agitation pendant 1 h. Le précipité vert olive obtenu a été récupéré par filtration, lavé à l'éthanol, à l'éther diéthylique, puis séché sous P4O10.
2.2.3 {Cu(H2LA)Br2} [C]
On a utilisé la même procédure que pour la préparation du complexe B. Nous avons pris CuBr2 (450 mg ; 2,01 mmol) au lieu de CuCl2·2H2O.
2.2.4 {Cu(LB)}2 [D]
Dans 50 ml de méthanol ont été dissous 247,7 mg (1 mmol) de H2LB et 83,92 mg (2 mmol) de LiOH·H2O. 241,6 mg (1 mmol) de Cu(NO3)2·3H2O, dissous dans 20 ml de méthanol, ont été ajoutés à la solution précédente. Le mélange obtenu est porté à reflux et maintenu sous agitation pendant 1 h. Le précipité formé est récupéré par filtration, lavé au méthanol, puis à l'éther, et séché sur P4O10.
Nous avons tenté de préparer, à partir de ce ligand, les complexes 1:1 avec CuCl2·2H2O, CuBr2, Cu(OAc)2·H2O, ainsi que le complexe 1:2, avec Cu(NO3)2·3H2O. Nous obtenons dans ces cas le complexe {Cu(LB)}2.
2.2.5 {Ni(LC)3/2H2O} [E]
356,4 mg (2 mmol) de H2LC ont été introduits dans 20 ml de méthanol ; 224,5 mg (4 mmol) de NaOH ont ensuite été ajoutés. La solution résultante a été agitée durant 30 min, puis chauffée à reflux pendant 1 h. Le précipité obtenu a été récupéré par filtration, puis lavé au méthanol et à l'éther, avant d'être séché sous P4O10.
3 Résultats et discussion
Les données infrarouge sont reportées dans le Tableau 2.
Données IR des complexes A, B, C, D et E
| A | B | C | D | E | Attribution |
| 1610 tF | 1630F, 1610 tF | 1630 F, 1610 tF | 1600 tF | 1600 tF | νCN [8–10] |
| 1520 tF | 1580 tF(large) | 1580 tF(large) | 1552 tF | νCO [8–10] | |
| 1570 f | 1540 tF | 1540 tF | νN–N [8–10] | ||
| 1260 tF | 1220 tF | 1220 tF | 1294 tF, 1271 tF | 1200 tF | νC–O [8–10] |
| 1383 tF | ν3(NO3−) ionique | ||||
| 1461tF, 1327 tF | ν3(NO3−) [7] | ||||
| 377 m | 353 f | 340 f, 327 F | 332 f | νM–N [7] |
Les complexes A, B et C ont été obtenus sans neutralisation préalable du ligand H2LA. Nous remarquons sur leurs spectres infrarouges les vibrations des principales bandes νCO (1580–1515 cm−1), νCN (1630–1610 cm−1), νC–O (1260–1220 cm−1). Pour le complexe D, nous retrouvons νCN à 1600 cm−1 et νC–O phénolique sous forme de doublet à 1294 et 1271 cm−1, alors que la bande νCO (E) est pointée à 1552 cm−1. Outre ces vibrations, les bandes relatives aux nitrates sont aussi observées. Sur le spectre IR de A, nous observons la bande due à ν3(NO3−) à 1461 et 1327 cm−1, alors que celle pointée à 1383 cm−1 est attribuée à un nitrate ionique [7]. L'éclatement de la ν3 en deux composantes indique un NO3− coordinant, de symétrie C2v ou Cs. Ces données IR montrent l'implication de l'azote de l'imine, de l'oxygène phénolique et de l'oxygène du groupement acétyl dans la coordination de l'ion métallique. Les vibrations νM–O et νM–N (Tableau 2), notées sur les spectres de basse fréquence, corroborent aussi ces résultats. Les spectres UV révèlent, en plus des transitions π → π∗ et n → π∗, des absorptions dues aux transitions d–d et aux transferts de charge métal–ligand (Tableau 3).
Données UV concernant les complexes A, B, C, D et E
| Composés | Longueurs d'onde | Attributions |
| A | 215 nm, 267 nm | π → π∗ |
| 310 nm | n → π∗ | |
| 400 nm | Transfert de charge (TC) | |
| 550–810 nm (absorption large) | d → d | |
| B | 210 nm, 250 nm | π → π∗ |
| 300 nm, 325 nm ep | n → π∗ | |
| 400 nm | Transfert de charge (TC) | |
| Absorption large jusqu'à 600 nm | d → d | |
| C | 260 nm | π → π∗ |
| 300 nm, 330 nm | n → π∗ | |
| 400 nm, 440 nm | Transfert de charge (TC) | |
| 500–1200 nm | d → d | |
| D | 265 nm | π → π∗ |
| 320 nm | n → π∗ | |
| 400 nm, 430 nm | Transfert de charge (TC) | |
| 470 nm, 730 nm | d → d | |
| E | Absorption large et intense jusqu'à 300 nm | π → π∗ et n → π∗ |
| 400 nm | Transfert de charge (TC) | |
| 500 nm, 850 nm | d → d |
Les mesures de susceptibilité magnétique du composé A à température variable dans le domaine (250–2 K) montrent que la susceptibilité croît avec l'abaissement de la température (Fig. 1a). Elle croît de façon plus nette à partir de 100 K, avant de tendre de façon asymptotique vers l'axe des ordonnées. Ce comportement ressemble à celui d'un centre paramagnétique obéissant à la loi de Curie. Le produit de la susceptibilité molaire par la température χM T décroît avec la température (Fig. 1b). Ce comportement est caractéristique des interactions antiferromagnétiques.
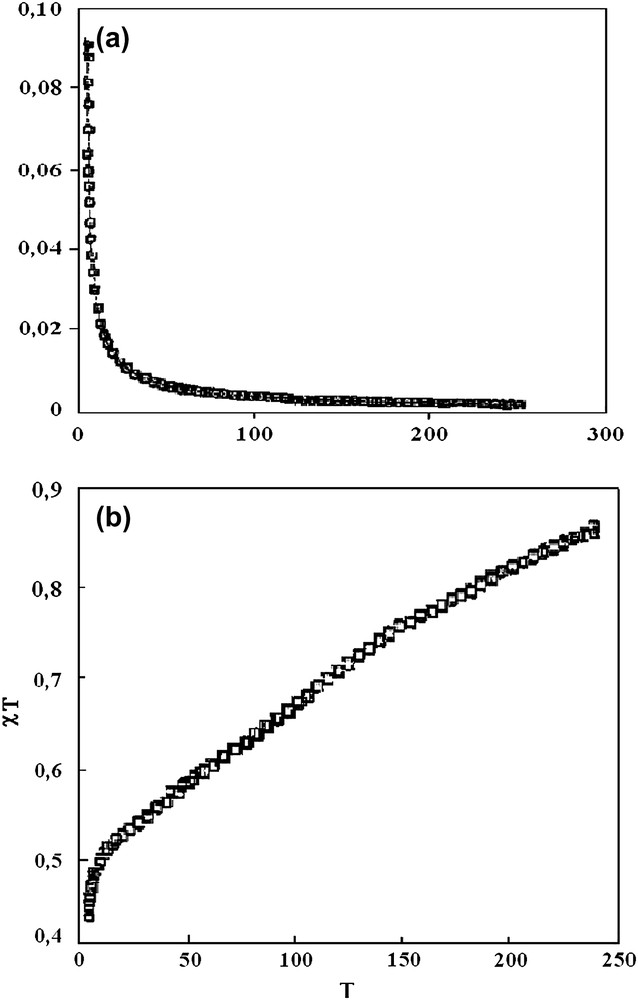
(a) Variation avec la température de la susceptibilité magnétique χM(T) du composé A. (b) Variation avec la température du produit χM T pour le composé A.
La valeur de χM T à la température ambiante de 0,88 cm3 K mol−1 pour le complexe A est proche de celle correspondant à deux ions Cu(II). Ainsi, une structure mononucléaire est dorénavant exclue. Cependant l'absence de maximum de χM T jusqu'à 3 K prouve que la constante de couplage J est faible.
Pour le complexe B, χM T tend vers une valeur de 0,17 cm3 K mol−1 à très basse température. En effet, une valeur faible de J est la conséquence d'un écart faible entre l'état fondamental singulet et l'état excité triplet. À la température limite, la population de spin de l'état excité est encore non négligeable ; ainsi, χM T est différent de zéro. Nous avons essayé de décrire le système en utilisant la relation de Curie–Weiss χM = C/(T − θ). Le tracé de la courbe 1/χM(T) permet d'obtenir deux segments de droite (Fig. 2a et b) dans le domaine de température compris entre 2 et 50 K (R = 1,49 ; déviation maximale = 3,62) et de 50 à 230 K (R = 0,86 ; déviation maximale = 2,37).
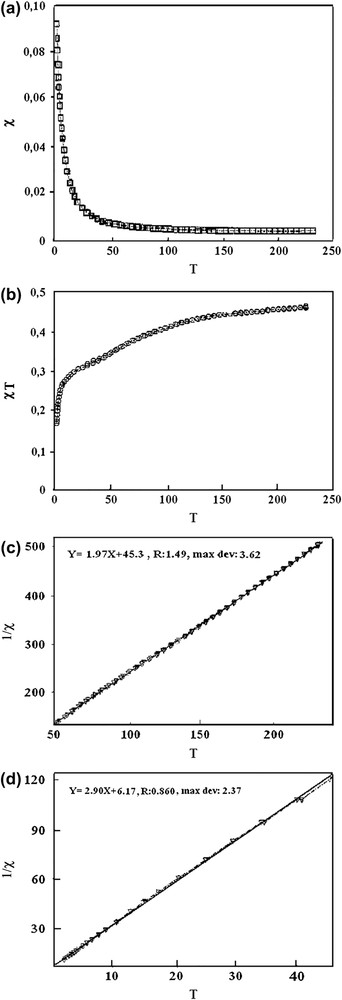
(a) Variation avec la température de 1/χ(T) pour le composé B dans le domaine 230–50 K. (b) Variation avec la température de 1/χ(T) pour le composé B dans le domaine 50–2 K. (c) Variation avec la température de la susceptibilité magnétique χM(T) du composé B. (d) Variation avec la température du produit χM T pour le composé B.
En extrapolant la courbe jusqu'à T = 0 K, nous obtenons θ = −2,12 K et C = 0,345 cm3 K mol−1. La relation θ = JS (S + 1)/3k donne une valeur de J = −5,89 cm−1, qui vérifie les interactions antiferromagnétiques faibles entre les deux centres magnétiques. Le complexe D, provenant de H2LB, a un comportement magnétique tout à fait différent. En effet, la courbe χM(T) (Fig. 3a) est, dans la région de 200 à 2 K, une droite parallèle à l'axe des abscisses à l'ordonnée χM = 0. Ceci indique que le composé est diamagnétique et que sa susceptibilité reste indépendante de la force du champ magnétique appliqué et de la variation de la température. Cependant, le produit de la susceptibilité par la température (Fig. 3b) décroît avec l'abaissement de la température, mettant ainsi en évidence une interaction antiferromagnétique très forte entre les centres magnétiques. Ces interactions sont tellement importantes que les deux électrons célibataires provenant des deux centres magnétiques se couplent pour former une paire. Ainsi, nous obtenons un ensemble compact sans électron non apparié, expliquant le caractère diamagnétique du complexe. Pour ces composés, des constantes de couplage J de l'ordre de −1000 cm−1 sont attendues [11]. L'état excité triplet se trouve à un niveau énergétique très élevé de l'état fondamental singulet. Ceci expliquerait la densité de spin presque nulle au niveau de cet état excité (S = 1), qui constitue l'état magnétique. À la température ambiante, seul l'état fondamental singulet (S = 0) non magnétique est peuplé ; le composé possède une susceptibilité invariable. L'étude des propriétés magnétiques du complexe E entre 250 et 2 K permet de distinguer deux domaines (Fig. 4a et b). Dans la région 250–100 K, la susceptibilité augmente légèrement avec l'abaissement de température, alors que le produit χM T est presque constant dans cet intervalle. La courbe 1/χM = f(T) est une droite, ce qui montre que le comportement magnétique du complexe est décrit dans ce domaine par la loi de Curie–Weiss (déviation maximale = 0,694 et R = 0,289). En dessous de 100 K, la susceptibilité croît très rapidement, avant de tendre de façon asymptotique vers l'axe des ordonnées aux basses températures. Le produit χM T décroît rapidement avec la température et prend une valeur de 0,17 cm3 K mol−1 à la température limite. La valeur de 0,96 cm3 K mol−1 obtenue pour χM T à la température ambiante montre que le complexe est mononucléaire et high spin. En effet, le nickel(II) est le centre magnétique. L'état fondamental triplet (S = 1) laisse prévoir un couplage spin orbite entraînant une variation assez importante de g par rapport à sa valeur pour l'ion libre. Cependant, ce phénomène ne semble pas être dominant pour le nickel, car le premier état excité se trouve à 800 cm−1 [12] ; il est, par conséquent, sans importance magnétique. Le nickel(II) se trouve dans un environnement octaédrique d'après les résultats spectroscopiques [13,14]. Dans une telle géométrie, l'état fondamental provenant de la configuration t2g6eg2 est 3A2 et les états excités qui découlent de t2g5eg3 sont 3T1 et 3T2. L'état excité de plus basse énergie est 3T2. Nous limiterons notre étude à cet état. Le couplage spin–orbite induit une levée de la dégénérescence à la suite de l'éclatement de 3T2 en 3A1 et 3E. Du fait de son énergie assez élevée par rapport au niveau fondamental, sa population de spin est pratiquement nulle. La décroissance rapide de χM T observée en dessous de 100 K ne saurait être liée à ce phénomène. Nous pensons plutôt à l'éclatement au champ nul zero-field splitting, combiné aux interactions antiferromagnétiques entre les différentes molécules monomères.
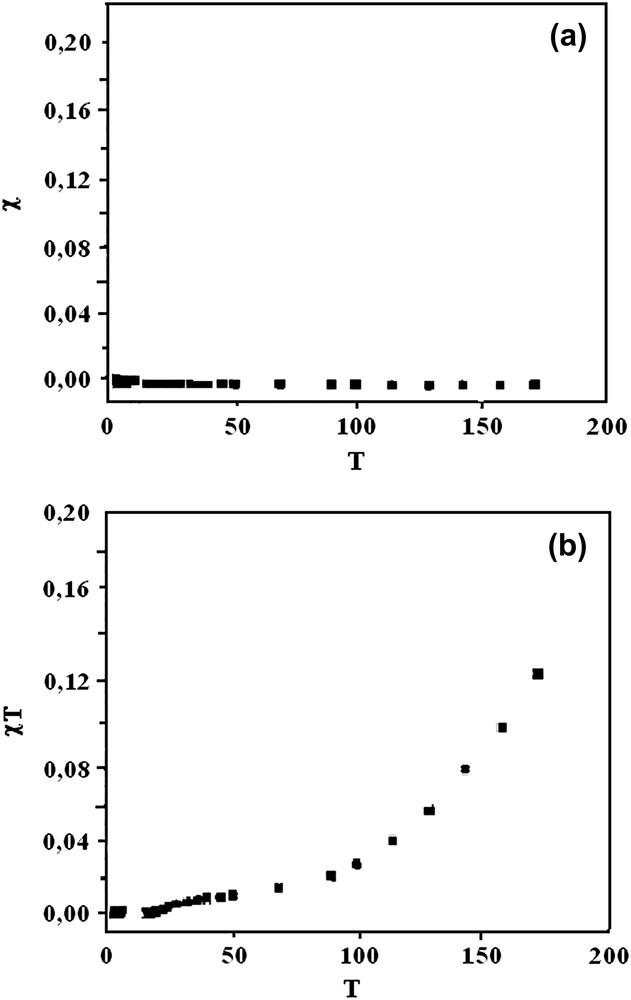
(a) Variation avec la température de la susceptibilité magnétique χM(T) du composé D dans le domaine 200–2 K. (b) Variation avec la température du produit χM T pour le composé D dans le domaine 200–2 K.
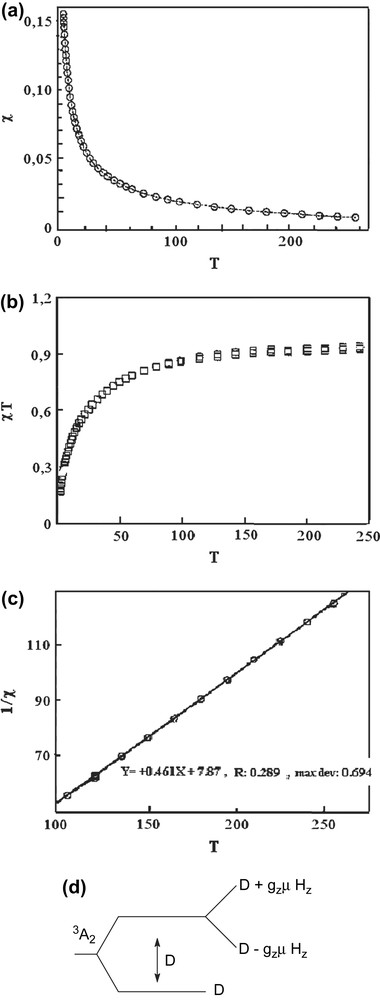
(a) Variation avec la température de la susceptibilité magnétique χM(T) du composé E dans le domaine 250–2 K. (b) Variation avec la température du produit χM T pour le composé D dans le domaine 250–2 K. (c) Variation avec la température de 1/χ(T) pour le composé E dans le domaine 250–100 K. (d) Dégénérescence de l'ion Ni (II).
L'éclatement au champ nul crée une anisotropie autour de l'ion Ni(II) ; l'application d'un champ magnétique extérieur permet de résoudre la dégénérescence, que nous illustrons par la Fig. 4d.
Le dépeuplement des niveaux d'énergie obéit à la loi de Boltzmann. Dans ce domaine de température, le dépeuplement des états excités découlant de l'éclatement au champ nul devient plus important, au profit de l'état fondamental. Il est aussi à considérer que ce phénomène seul ne peut pas justifier l'allure de χM T(T). Les molécules situées dans un même plan ou dans des plans voisins sont en interaction, soit de façon électrostatique, soit par liaison de faible énergie, de façon à conférer à l'édifice une nature antiferromagnétique. Falvello et al. [15] ont obtenu des résultats similaires dans le cadre de leurs travaux sur les propriétés magnétiques des cyanurates N du nickel(II). Ils ont déterminé, à l'aide des rayons X, l'existence de liaisons hydrogène entre les différents motifs.
L'étude électrochimique par voltamétrie cyclique des complexes A, B et D est effectuée en solution dans l'acétonitrile, le DMF ou le THF. Les électrolytes supports sont le tétrafluoroborate de tétrabutylammonium et le perchlorate de tétrabutylammonium. Le complexe à étudier est dissous dans la solution contenant le sel et les mesures sont effectuées dans le domaine d'électroactivité déjà défini.
Nous avons représenté sur les Fig. 5a et b les voltamogrammes du composé D. Nous avons reporté dans le Tableau 4 l'ensemble des potentiels redox ainsi que les valeurs de la constante de coproportionnalité des produits A, B et D. L'ensemble des courbes révèle deux vagues en réduction et deux vagues en oxydation, indiquant un processus d'oxydoréduction en deux étapes, avec des échanges monoélectroniques successifs (Figs. 6–9) :
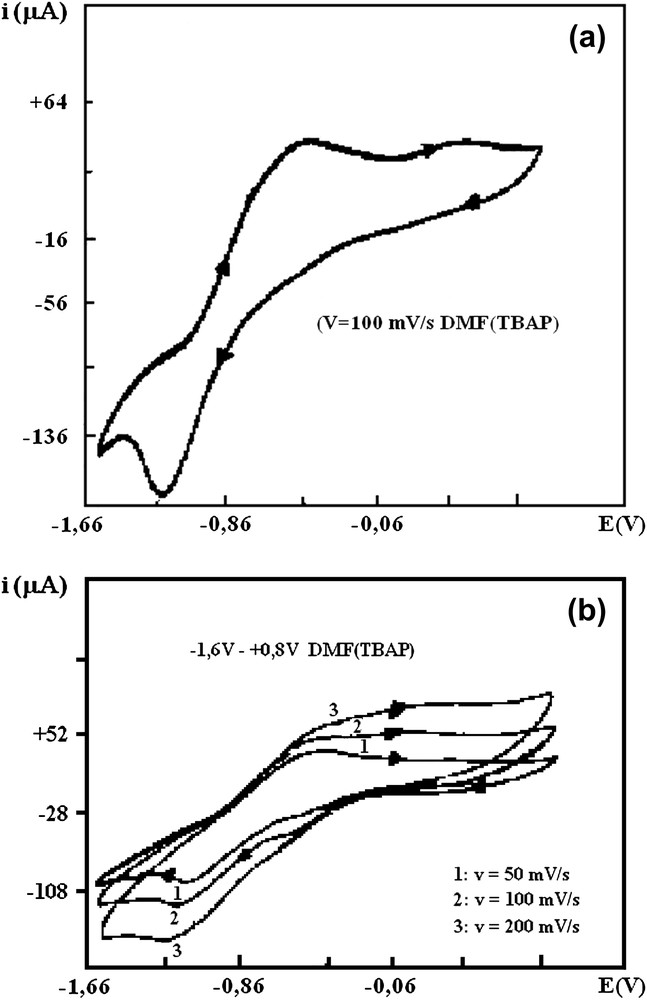
(a) Voltamogramme du composé D (−1,6 V → +0,8 V), DMF(TBAP). (b) Voltamogrammes du composé D à différentes vitesses de balayage (−1,6 V → +0,8 V), DMF(TBAP).
Données électrochimiques concernant les composés A, B et D
| Composé | solvant | Ered(V) | Eox(V) | ΔE | Kco | ||
| A | CH3CN | −0,5 | −0,3 | −0,4 | −0,85 | −0,45 | 4,08 × 107 |
| −1,1 | −0,6 | ||||||
| THF | −0,5 | −0,2 | −0,35 | −0,75 | −0,4 | 5,82 × 106 | |
| −0,9 | −0,6 | ||||||
| B | DMF | −0,6 | +0,2 | −0,2 | −0,75 | −0,55 | 2 × 109 |
| −0,9 | −0,6 | ||||||
| D | DMF | −0,6 | −0,1 | −0,2 | −0,55 | −0,35 | 8,30 × 106 |
| −1,1 | −0,6 |
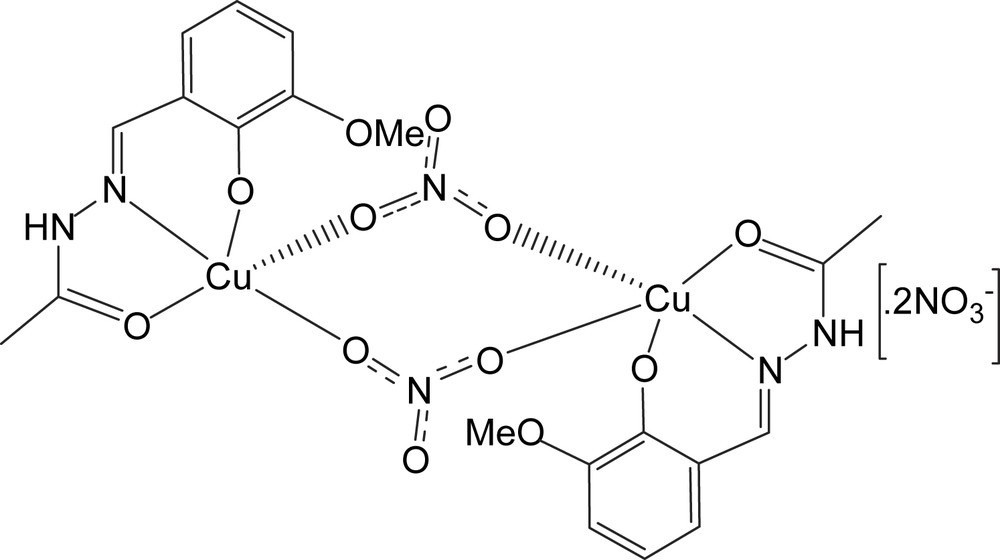
Structure du composé A.
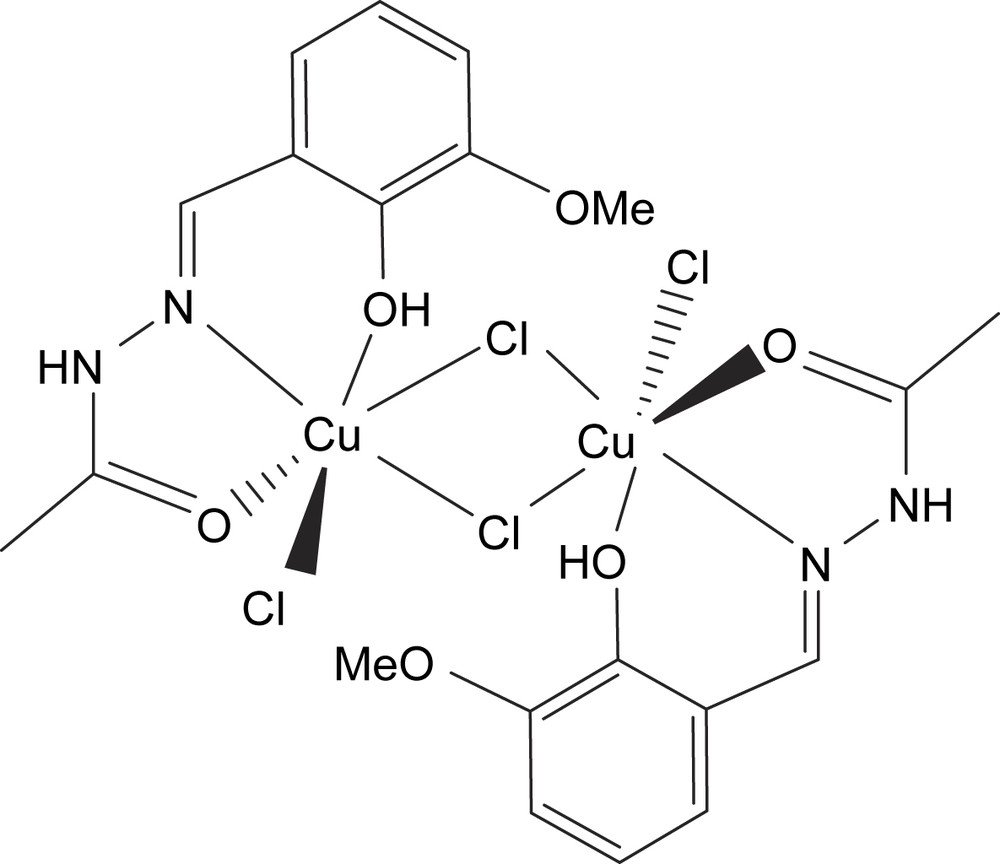
Structure du composé B.
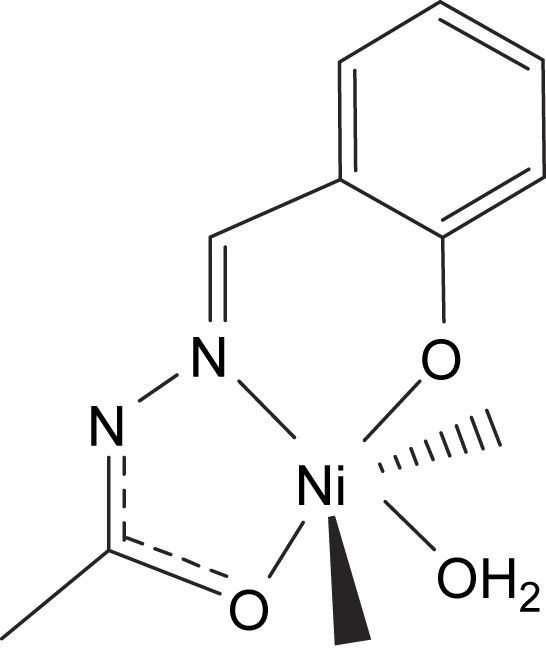
Structure du composé C.
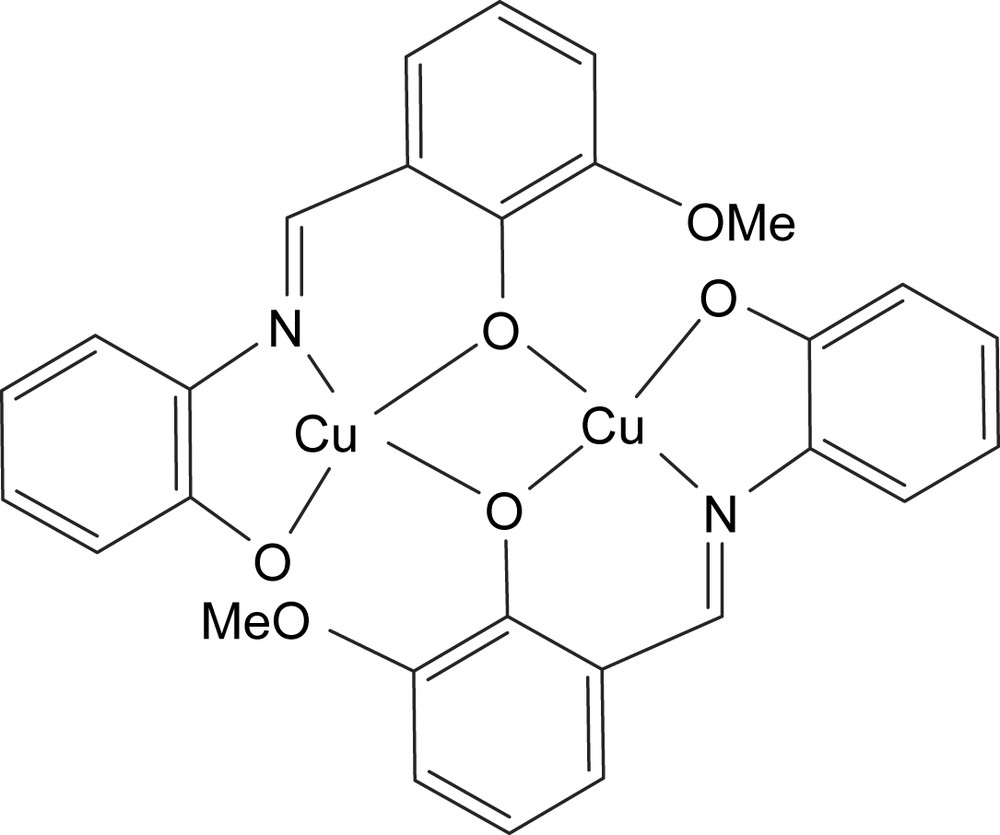
Structure du composé D.
Ces processus en deux étapes ont été déjà rencontrés dans nos études antérieures sur les complexes homo- et hétérobinucléaires du 2,6-bis (carboxyéthylthiométhyl)-4-méthylphénol [16] ainsi que dans la littérature [17–24]. Nous pouvons conclure à une interaction entre les deux centres métalliques, confirmant ainsi la dimérisation déduite des mesures magnétiques. Les processus rédox envisagés sont quasi réversibles. Ceci est attesté par les valeurs de Ep − Ep½ (différence entre le potentiel de pic Ep et le potentiel de demi-pic Ep½). De même, le rapport ipa/ipc (ipa = courant de pic anodique, ipc = courant de pic cathodique) est différent de 1 [25]. Les valeurs de Kco reportées dans le Tableau 4 sont relativement élevées. Elles sont toutes supérieures à 106, ce qui permet de les classer parmi les systèmes complètement délocalisés (classe III) [26]. Elles démontrent la stabilité de l'espèce intermédiaire CuIICuI(Li)2. En faisant un balayage cyclique, nous notons une disparition progressive du premier pic de réduction en faveur du deuxième, qui devient plus important. Cependant, nous observons deux pics nets en oxydation, qui ne peuvent correspondre qu'à deux réductions, qui se sommeraient en une vague, d'où l'importance de celle-ci. En augmentant la vitesse de balayage de 20 à 200 mV s−1 (Fig. 5a et b), nous remarquons que le nombre de pics reste inchangé, de même que l'allure générale des courbes. Nous pouvons donc conclure à une absence de décomplexation ou de décomposition de nos composés [27].
Nous remarquons cependant un seul pic d'oxydation et un seul pic de réduction pour le complexe D à la vitesse de 500 mV s−1. Nous supposons l'existence d'effets cinétiques. Lorsque la vitesse de balayage est faible (20 et 50 mV s−1), les deux pics d'oxydation et de réduction deviennent nets. Le courant de pic varie linéairement par rapport à la racine carrée de la vitesse de balayage, ce qui prouve qu'on est en présence d'un processus de diffusion.
4 Conclusion
Ce travail nous a permis de confirmer la formation des ligands H2LA, H2LB et H2LC à la suite de la condensation de l'acétylhydrazine ou du 2-aminophénol avec le salicylaldéhyde ou l'o-vanilline. La spectroscopie infrarouge montre l'implication des oxygènes phénolique et acétylique ainsi que de l'azote de l'imine dans la coordination des ions métalliques.
Les mesures magnétiques révèlent des interactions de nature antiferromagnétique pour les complexes dinucléaires du cuivre(II).
La voltamétrie cyclique a permis d'élucider des processus d'oxydoréduction quasi réversibles en deux étapes, avec des échanges monoélectroniques successifs.
Remerciements
Les auteurs remercient l'Agence universitaire de la francophonie pour son soutien financier dans le cadre du projet de coopération interuniversitaire (AUF-PSCI n° 6301PS48).